
ONCOLOGÍA
INTRODUCCIÓN A LAS NUEVAS PERSPECTIVAS EN LA DECODIFICACIÓN DEL GENOMA TUMORAL
La validación clínica del perfil genómico completo del cáncer en la atención oncológica se ha logrado a través de la demostración de resultados ampliamente mejorados; actualmente los avances en la decodificación genómica del cáncer es un área activa de investigación cuyos resultados han comenzado a beneficiar el éxito del tratamiento en pacientes con cáncer. Sin embargo, la adopción de tecnología en un contexto clínico, como una prueba de rutina para apoyar la selección de la terapia para pacientes con cáncer, enfrenta múltiples desafíos.
Es importante mencionar estos desafíos para la obtención y análisis de tejido neoplásico, se resumen en tres premisas principales: 1) La mayoría de las muestras de cáncer suelen procesarse mediante técnicas que pueden dañar el ácido desoxirribonucleico (ADN), además pueden conservarse por medio de fijado con formol e inclusión en parafina, por lo que se deben aplicar protocolos robustos de extracción de ADN y secuenciación. 2) Muchas muestras disponibles para pruebas son biopsias con aguja fina o bloques celulares preparados a partir de derrames pleurales malignos, pericárdicos o peritoneales, que requieren protocolos que acomodan cantidades limitadas de tejido y ADN extraído. 3) Mientras que la selección de muestras con alto contenido tumoral es factible en el entorno de la investigación, se debe lograr un perfil preciso en las muestras clínicas cuando la proporción relativa de células tumorales por muestra es baja.
Pensando en esta gran problemática, científicos han desarrollado y validado una prueba de perfil del genoma del cáncer basada en la "secuenciación de última generación" (NGS, por sus siglas en inglés) que interroga 4 557 exones de 287 genes relacionados con el cáncer y ha establecido puntos de referencia de rendimiento que apoyan el uso clínico directo. Evaluando la sensibilidad analítica, la especificidad y la precisión en todo el rango informable del ensayo, con una representación de los tipos de muestra más relevantes en cáncer fijadas e incluidas en parafina.
Mediante la prueba de sustituciones de bases, indeles, amplificaciones de genes focales y deleciones de genes homocigotos, Frampton, et al. lograron un análisis de 2 221 muestras fijadas e incluidas en parafina (MFP) de tumores de pacientes enviadas al laboratorio de patología acreditado por el Colegio de Patólogos Americanos, lográndose con éxito proporcionar información importante y relevante para la incorporación exitosa del sistema de NGS en la práctica oncológica de rutina y en ensayos clínicos. En la imagen que a continuación se presenta, se puede apreciar la metodología a seguir por el NGS para la detección de anormalidades genéticas en el cáncer.
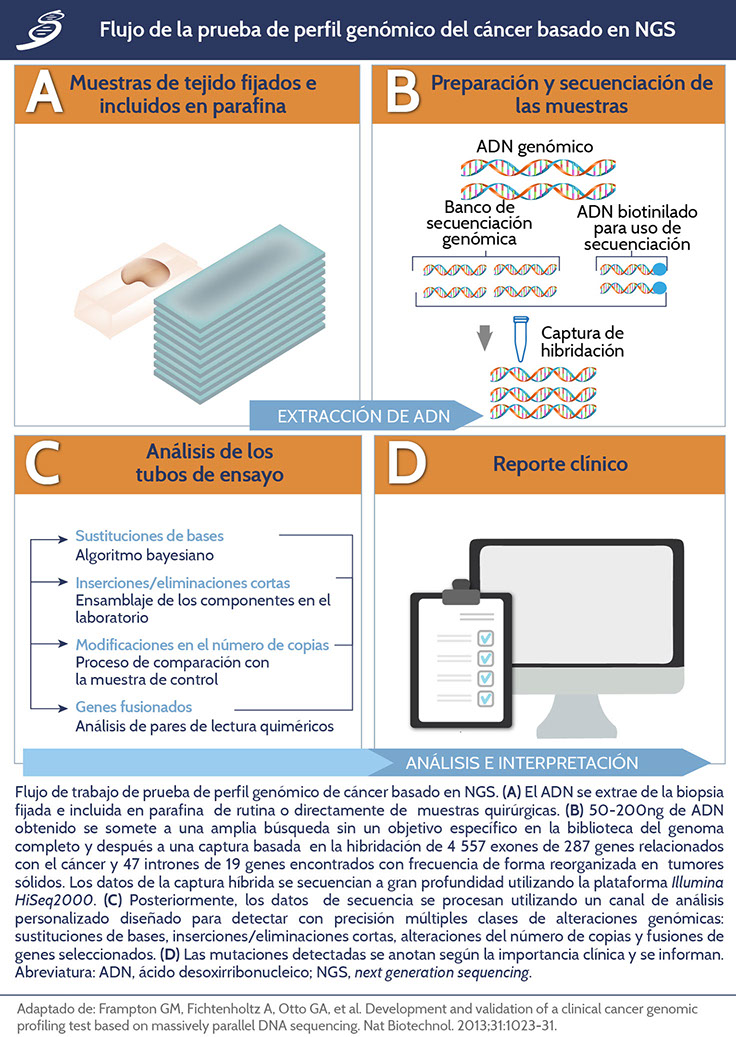
En general, las muestras clínicas de cáncer obtenidas en el curso de la atención de rutina parecen susceptibles de un análisis de diagnóstico optimizado basado en NGS. Los informes se centran principalmente en sitios conocidos de mutación somática, truncamientos o deleciones homocigóticas de genes supresores de tumores conocidos, así como amplificaciones conocidas de oncogenes y fusiones génicas que se han reorganizado en tumores sólidos.
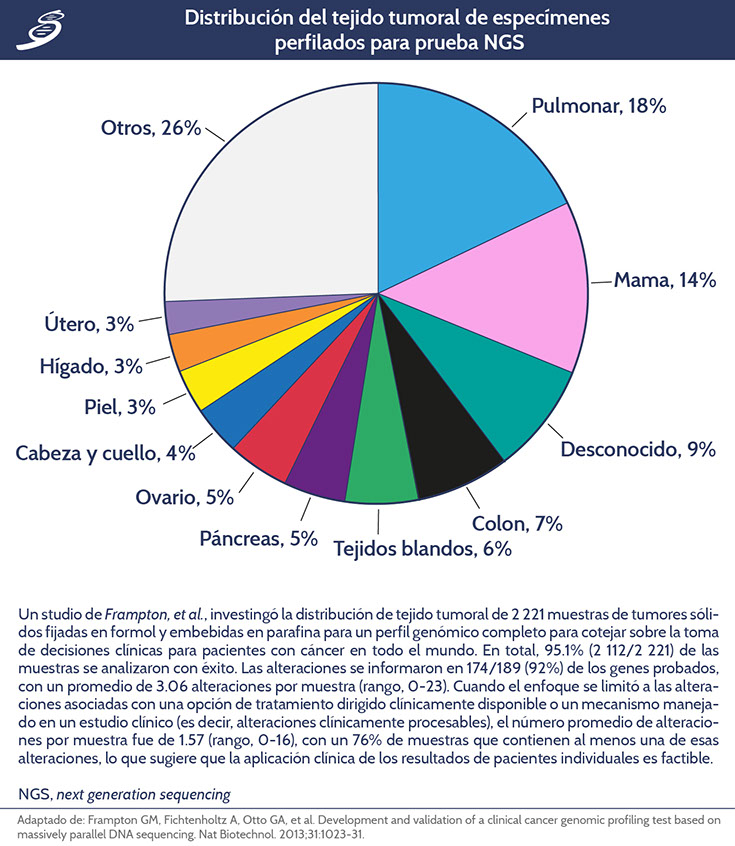
De acuerdo a la prueba de Fampton, las alteraciones se informaron en 174/189 (92%) de los genes probados, con un promedio de 3.06 alteraciones por muestra (rango, 0-23), sin embargo, cuando el enfoque se limitó a las alteraciones asociadas con una opción de tratamiento dirigida clínicamente disponible o un ensayo clínico impulsado por un mecanismo (es decir, clínicamente accionable), el número promedio de alteraciones por muestra fue de 1.57 (rango, 0-16), con un 76% de muestras que contienen al menos una de esas alteraciones, lo que sugiere que la aplicación clínica de los resultados de pacientes individuales es factible.
Aunque el número de alteraciones en la muestra de cualquier paciente con cáncer individual fue bajo (promedio, 1.57), se observó una gran variedad de alteraciones en todas las muestras, con 1 579 alteraciones únicas informadas. Por lo tanto, no fue sorprendente observar que los paradigmas actuales de pruebas clínicas, que comprenden sólo puntos críticos de mutación, capturan menos de un tercio del total de resultados en alteraciones.
Las implicaciones terapéuticas son particularmente en terapias dirigidas, como es el caso de HER2 (receptor 2 del factor de crecimiento epidérmico humano). Aunque actualmente HER2 está validado clínicamente sólo como un objetivo farmacológico amplificado o sobreexpresado en cáncer de mama y gastroesofágico, observamos alteraciones de HER2 en 12 tipos de tumores sólidos adicionales que abarcan la mayoría de los sitios conocidos de activación en todo el gen y que comprende el 5% del total de casos. Es importante destacar que más del 40% de todas las alteraciones HER2 fueron mutaciones puntuales, inserción o deleción (también denominado “indel”) en muestras no amplificadas que habrían sido negativas en otras pruebas de biomarcadores limitados.
El tratamiento del cáncer ha entrado en una nueva era en la que los diagnósticos sofisticados revelan las causas moleculares para la aparición de tumores en pacientes de forma individual, ofreciendo la oportunidad de seleccionar una terapia dirigida apropiada. Sin embargo, a medida que se identifican más genes de cáncer clínicamente relevantes, hacer coincidir a los pacientes con la terapia dirigida óptima se vuelve más desafiante, particularmente porque la práctica clínica rutinaria presenta obstáculos que normalmente no se encuentran en el entorno de la investigación.

Es importante destacar que la mayoría de las muestras clínicas son MFP, las cuales pueden dañar el ADN y requieren metodologías optimizadas para analizarse con precisión. Las biopsias que son tomadas en neoplasias avanzadas, están asociadas con mayor morbilidad y obtienen muestras más pequeñas en comparación de las biopsias con aguja fina en otras neoplasias, lo que requiere el desarrollo de metodologías que puedan detectar todas las clases de alteración genómica en un solo tiempo diagnóstico para la preservación de tejido. Además, la selección de tumores con el mayor porcentaje de núcleos tumorales, práctica común en el entorno de investigación, no es una opción clínica. En muestras de neoplásicas con un bajo porcentaje de tejido tumoral (altos niveles de contaminación de otras células), se requiere una prueba de sensibilidad alta. También se requiere una alta especificidad, ya que los resultados falsos positivos pueden llevar a una elección terapéutica subóptima.
Finalmente, las alteraciones genómicas que son individualmente raras, pero que acumulativamente forman una fracción sustancial de alteraciones genómicas clínicamente y biológicamente relevantes, requieren el análisis de miles de exones y de cientos de genes relacionados con cáncer para maximizar las opciones de tratamiento específicas, sin embargo, la prueba diagnóstica basada en NGS para detectar con precisión todas las alteraciones genómicas clínicamente relevantes en todos los exones de codificación de 287 genes de cáncer en muestras clínicas de MFP de rutina, incluidas las biopsias con aguja, hasta ahora muestran grandes resultados.
1. Frampton GM, Fichtenholtz A, Otto GA, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31:1023-31.
2. Foundation Medicine. Genomic Testing. Comprehensive Genomic Profiling [2017]. Consultado en: https://www.foundationmedicine.com/ [Revisado en enero 2018].
Periodismo científico cultural
Sobre el cigarro electrónico
Ya ha pasado tiempo desde que el cigarro se convirtió en el enemigo público número uno, tanto para fumadores como para no fumadores...
[leer mas]
la sal! Tu cerebro
y corazón lo agradecerán
La enfermedad diverticular y sus complicaciones son una causa importante de morbimortalidad en todo el mundo...
[leer mas]
El primer anticuerpo monoclonal para pacientes con hemofilia A e inhibidores a FVIII
Cuando se vive con hemofilia todo puede ser una amenaza para provocar sangrados; desde niños...
[leer mas]
Infografías




TODOS LOS DERECHOS RESERVADOS © SPG COMUNICACIONES SA DE CV 2012-2018
Diseño: A. Victoria Pérez
Aviso de privacidad