__
__
__
EDUCACIÓN MÉDICA
SÍNDROME CORONARIO AGUDO
FISIOPATOLOGÍA
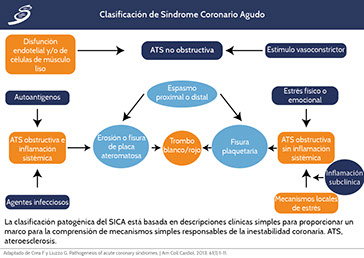
Los mecanismos básicos responsables de la inestabilidad coronaria en síndrome coronario (SICA) se pueden —para fines prácticos— dividir de acuerdo a su etiología básica:
• Aterosclerosis obstructiva e inflamación sistémica
• Aterosclerosis obstructiva sin inflamación sistémica
• Sin aterosclerosis obstructiva
SICA SECUNDARIO A ATEROESCLEROSIS Y RESPUESTA INFLAMATORIA SISTÉMICA
Complicaciones trombóticas de la ateroesclerosis
La erosión superficial de una arteria coronaria a través de la capa íntima de un nódulo calcificado que tiene una hemorragia interna en una placa ateromatosa desencadena un porcentaje de los SICA.
Una capa fibrosa suele superponerse al centro de la placa ateromatosa rica en lípidos, también conocida como núcleo necrótico (como se muestra en la siguiente imagen). Esta cápsula fibrosa se encuentra entre el compartimiento sanguíneo, con sus factores de coagulación latentes, y el núcleo lipídico, una porción de la placa llena de material trombogénico; tales placas a menudo, pero no siempre, tienen finas cápsulas fibrosas (50 a 65 μm de grosor, causa de estenosis luminal). Las placas rotas también tienden a presentar grandes núcleos lipídicos y abundantes células inflamatorias, así como puntilleo o calcificaciones irregulares; típicamente, los sitios donde las placas se rompen y provocan eventos coronarios fatales tienen pocas células musculares lisas.
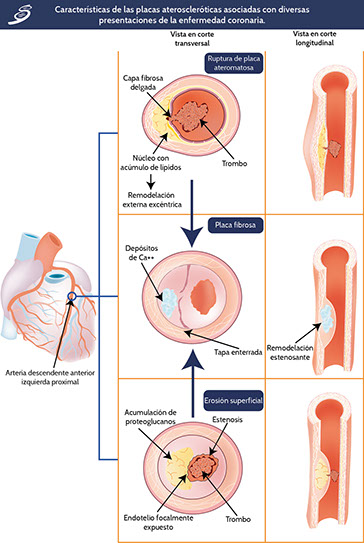
Vías inflamatorias predisponentes a la ruptura y trombosis de las arterias coronarias
Cuando se rompe la tapa fibrosa de una placa ateromatosa permite que la sangre entre en contacto con el material trombogénico del núcleo lipídico que puede producir trombosis. Cuando esto sucede, el factor tisular, un potente procoagulante producido por macrófagos en el núcleo de la placa, desencadena la cascada de la coagulación con generación y activación de trombina con posterior agregación plaquetaria. La misma señal proinflamatoria que aumenta la producción de colagenasa (CD154), también induce la expresión del factor tisular en los fagocitos mononucleares (de acuerdo a la imagen siguiente de ateroesclerosis con respuesta inflamatoria). Así, las células inflamatorias y los mediadores no sólo regulan la síntesis y la descomposición del colágeno, sino que también aumentan el potencial trombogénico de la placa aterosclerótica.
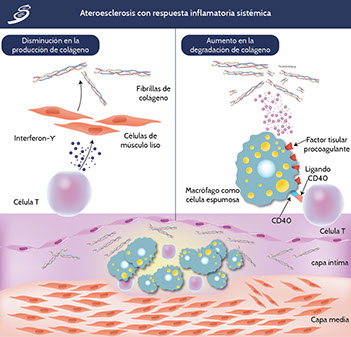
Una sección transversal de una placa ateromatosa en la parte inferior de la imagen previa muestra el núcleo central de lípidos que contienen células espumosas (antes macrófagos [amarillo]) y linfocitos T (azul). La capa íntima y media de los vasos también contiene células musculares lisas arteriales (rojas), que son la fuente del colágeno arterial (representado como estructuras enrolladas helicoidales triples). Las células T activadas (subtipo T helper tipo 1 [Th-1]) segregan interferón-γ, que inhibe la producción del nuevo colágeno intersticial que es necesario para reparar y mantener el tapón fibroso protector de la placa (imagen anterior,parte superior izquierda). Las células T también pueden activar a los macrófagos causando una lesión en la capa íntima de los vasos mediante la expresión del ligando CD40 (CD154), el cual compromete su receptor afín (CD40) en el fagocito. Esta señalización inflamatoria provoca la sobreproducción de colagenasas intersticiales (metaloproteinasas de matriz [MMPs] 1, 8 y 13) que catalizan el aumento en la velocidad de la degradación del colágeno (imagen previa, arriba a la derecha). El ligando CD40 también causan una sobreproducción de factor tisular procoagulante de los macrófagos. Por lo tanto, la señalización inflamatoria causa un tapón fibroso defectuoso por la disminución en la síntesis de colágeno y aumento de su ruptura al estar defectuoso, haciendo una placa susceptible a la ruptura. La activación inflamatoria también aumenta la producción de factores tisulares, lo que desencadena la formación de trombos en la placa fisurada. Estos mecanismos inflamatorios pueden precipitar complicaciones trombóticas, incluyendo los SICA.
(Véase también Procesos biológicos centrales para la patogénesis de aterosclerosis en Infarto agudo al miocardio con elevación del segmento ST)
SICA secundario a ateroesclerosis sin respuesta inflamatoria
Los mecanismos etiológicos incluyen situaciones emocionales externas de gran impacto para el paciente; suelen ser eventos de corta duración o incluso manifestaciones agudas de disposiciones emocionales internas más duraderas. Aparte de las anteriores, un esfuerzo físico intenso y estrés mecánico local a nivel de la pared arteriolar, es decir, esfuerzo circunferencial de la arteria y estrés por cizallamiento.
Además, la inflamación subclínica en el microambiente de la estenosis se cree que tiene un papel importante en la cadena de eventos que conducen a la inestabilidad coronaria, aunque el desencadenante y los mecanismos de inflamación son probablemente diferentes de los que operan en pacientes con evidencia sistémica de inflamación. Otra etiología causal de inestabilidad coronaria es el espasmo coronario, el cual ha evidenciado en angiografías coronarias (estudio CASPAR) espasmo coronario en un 50% de la población de estudio secundario a la administración intracoronaria de acetilcolina, por lo que puede o no coexistir en presencia de placa ateroesclerótica obstructiva.
SICA sin ateroesclerosis obstructiva
En ausencia de aterosclerosis obstructiva, las alteraciones funcionales de las arterias coronarias epicárdicas o de la microcirculación coronaria son la causa probable de inestabilidad y SICA. Fisuras de placa ateromatosa se han observado en estudios recientes de mujeres con SICA y sin aterosclerosis obstructiva, lo que se cree sería la causa de formación transitoria de trombos. Sin embargo, su función patógena es difícil de establecer porque la fisura de la placa es con frecuencia asintomática y se observa en una proporción considerable de pacientes con cardiopatía isquémica estable.
La constricción intensa de la microcirculación coronaria es otro mecanismo que puede causar SICA en pacientes con ateroesclerosis coronaria no obstructiva. Este es el mecanismo probable de inestabilidad en pacientes con síndrome de Takotsubo, que se caracteriza por dolor isquémico en reposo, elevación del segmento ST, liberación de biomarcadores cardiacos y una acinesia regional característica que afecta más frecuentemente regiones distales del miocardio asociadas con hipercontractilidad de las regiones restantes.
Se ha encontrado el genoma del parvovirus B19 en biopsias miocárdicas de pacientes con arterias coronarias angiográficamente normales que presentan dolor isquémico típico, cambios en el segmento ST isquémico, aumento de troponinas y diversos grados de anomalías regionales del movimiento de la pared, esto se asoció con vasoconstricción coronaria en respuesta a la acetilcolina, lo que sugiere una disfunción endotelial severa.
Finalmente, la constricción microvascular de menor intensidad que se produce en ausencia de anomalías regionales del movimiento de la pared y asociada con un patrón inestable de angina, cambios en el segmento ST isquémico y/o aumento de troponinas, ha sido propuesta como contraparte microvascular de angina inestable,más frecuente en mujeres que en hombres.
Bibliografía
1. Mayo Clinic Staff. Acute Coronary Syndrome, MAYO CLINIC, [updated May 06, 2016] Disponible en: http://www.mayoclinic.org/diseases-conditions/acute-coronary-syndrome/home/ovc-20202307 [Revisado julio 2017]
2. Pries AR, Badimon L, Bugiardini R, Camici PG, Dorobantu M, Duncker DJ, et al. Coronary vascular regulation, remodelling, and collateralization: mechanisms and clinical implications on behalf of the working group on coronary pathophysiology and microcirculation, Eur Heart J, 2015; 36(45):3134-46.
3. Crea F y Liuzzo G. Pathogenesis of acute coronary síndromes, J Am Coll Cardiol, 2013; 61(1):1-11.
4. Libby P. Mechanisms of acute coronary syndromes and their implications for therapy, N Engl J Med, 2013; 368(21):2004-13.
Thygesen K, Alpert JS, Jaffe AS, Simoons ML, Chaitman BR, White HD, et al. Third universal definition of myocardial infarction, Circulation, 2012;126:2020-2035.
5.Timmis A. Acute coronary síndromes, BMJ, 2015; 351:h5153.
Aviso de privacidad
Diseño: A. Victoria Pérez