__
__
__
EDUCACIÓN MÉDICA
PeriodoNeonatalSíndrome de aspiración de meconioEnfermedad hemolíticadel recién nacidoFormación de membrana Hilaína
SÍNDROME DE DIFICULTAD RESPIRATORIA:
FORMACIÓN DE MEMBRANA HIALINA
FISIOPATOLOGÍA
El síndrome de dificultad respiratoria en el recién nacido se caracteriza por signos y síntomas comunes, pero de múltiples etiologías; las causas en las que está implicado el sistema respiratorio podrían categorizarse en: obstrucción aérea, enfermedad del parénquima pulmonar, malformaciones congénitas (causa anatómica) y síndromes de fugas aéreas. Las patologías tienen en común la lesión pulmonar e inflamación, con una deficiencia secundaria de factor surfactante que resulta en un mecanismo alterado de ventilación/perfusión (V/Q).
No existe una prueba de laboratorio como tal para el diagnóstico de síndrome de dificultad respiratoria, por lo que el diagnóstico será clínico y con auxiliares diagnósticos; la historia natural del padecimiento termina en enfermedad de la membrana hialina. Para este tema se analizará la enfermedad de la membrana hialina en recién nacidos de pretérmino (RNPT) que, por definición, son aquellos que nacen antes de las 37 semanas de gestación (SDG). En los RNPT es usual la aparición de síndrome de dificultad respiratoria (SDR) debido a la deficiencia relativa de surfactante que, al combinarse con la pobre distensibilidad del neonato, promueve atelectasias alveolares.
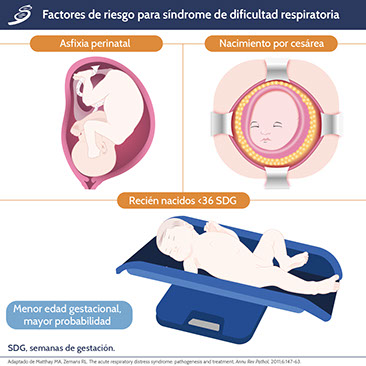
Otros factores de riesgo para el desarrollo de SDR incluyen diabetes materna; sepsis de inicio temprano; y, menos común, deficiencia congénita de surfactante.
Factor surfactante
La producción de surfactante comienza entre las 24 y 28 SDG y no es completamente funcional hasta mínimo 35 SDG. Durante las 34 y 36 SDG es que se lleva a cabo la síntesis y producción de surfactante por neumocitos maduros tipo II, además de alveolización de los sáculos alveolares que se convierten en alveolos y neumocitos tipo I maduros.
En el periodo comprendido entre las 34 y 36 semanas de gestación, durante la alveolarización, los sáculos alveolares se convierten en alvéolos maduros con neumocitos maduros tipo I y tipo II, siendo los últimos responsables de la síntesis y producción de surfactante.
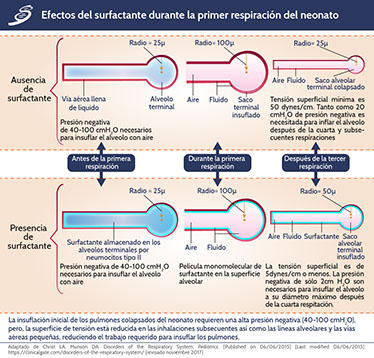
El factor surfactante es un ensamblado complejo de lípidos y proteínas producidas por neumocitos tipo II, los cuales lo secretan como una capa delgada de líquido que recubre la superficie alveolar que estará en contacto con el aire. De manera simultánea, desempeñará un papel defensivo como barrera contra patógenos y un estabilizador de la interfaz expuesta al aire que, de manera biofísica por sus propiedades lipídicas, impide el colapso alveolar disminuyendo la tensión superficial.
Los lípidos del surfactante forman alrededor del 90% del mismo, el otro 10% lo constituyen proteínas. La fracción lipídica se compone de un alto porcentaje de fosfolípidos (80-85%) y de un 5-10% de lípidos neutros, de los cuales, 75% de los fosfolípidos son fosfatidilcolinas. El dipalmitoilfosfatidilcolina (DPPC) es el fosfolípido responsable de la capacidad tensioactiva del surfactante para producir una tensión superficial extremadamente baja al final de la espiración, debido al alto contenido en el empaquetamiento de moléculas de DPPC desaturado que pueden sostenerse luego de una compresión. Además, al tener una carga negativa, su interacción con proteínas hidrofóbicas catiónicas es indispensable para la formación de una superficie activa tensioactiva.
Inflamación en el síndrome de dificultad respiratoria
El síndrome de dificultad respiratoria tiene una relación inversamente proporcional a la edad gestacional. Clínicamente puede ser difícil la distinción entre el síndrome y la taquipnea transitoria del recién nacido, que, como se menciona, forman parte del mismo espectro de enfermedad respiratoria de origen en el sistema respiratorio, que son las más comunes, tanto para RNPT, como en recién nacidos a término.
El síndrome de dificultad respiratoria tiene una patogénesis secundaria al proceso inflamatorio que se desarrolla asociándose así a una lesión pulmonar, como se ha dicho, independientemente del origen. La cascada inflamatoria que desencadena el daño puede ser pulmonar o sistémica, mediando la lesión alveolar por medio de las células del sistema inmune, además de lesión epitelial y vascular.
En el alveolo, los macrófagos secretan citocinas, interleucinas (IL) como IL-1, IL-6, IL-8 e IL-10 y factor de necrosis tumoral alfa (TNF-α) que de manera local estimulan la quimiotaxis para la activación de neutrófilos, los cuales son responsables de la liberación de especies reactivas de oxígeno (ROS), proteasas, leucotrienos, metaloproteinasas y factor activador de plaquetas (PAF); dichas sustancias amplifican el daño epitelial y endotelial afectando células alveolares tipo I y II. De esta manera se incrementa la permeabilidad al espacio alveolar conduciendo a una fuga amplificada de fluido (fuga) capilar. Este proceso terminará en un edema inflamatorio pulmonar masivo y formación de la membrana hialina, como se muestra a continuación en el.
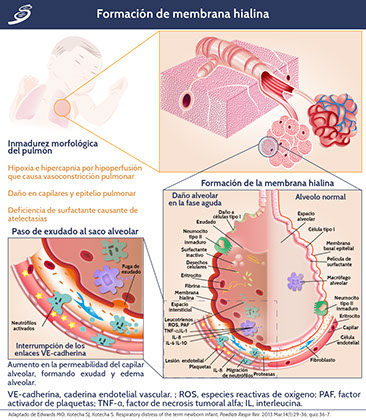
Membrana hialina y lesión pulmonar
Además de la reducción severa de surfactante en los RNPT, la lesión del parénquima pulmonar es también responsable de la inactivación de los complejos del surfactante restante. Proteínas plasmáticas como albúmina y fibrinógeno, compiten con el surfactante pulmonar para acomodarse en la interfaz de aire-agua. Dichas proteínas forman una película en la superficie alveolar que crea una barrera estérica y de energía electrostática que impide la incorporación de los fosfolípidos del surfactante en la interfaz, además, los fosfolípidos son insolubles y alcanzan la interfaz mediante un proceso de difusión cooperativa de grandes agregados moleculares, seguido del desempaque, reestructuración de la membrana y difusión de la monocapa. Por lo tanto, debido a que los fosfolípidos se transportan más lento a la interfaz, a menudo no puede desplazar las otras proteínas para formar una película activa en la superficie alveolar.
La lesión pulmonar causa un aumento en la degradación del surfactante, disminuyendo su concentración, y así, con la falta de reemplazo y la dilución en el líquido edematoso que se filtra de capilares a los alveolos, los mismos no pueden alcanzar tensiones superficiales mínimas suficientemente bajas durante la exhalación, debido a la compresión ineficiente, llevando al colapso alveolar y alteración de la oxigenación por shunt.
Consecuencias metabólicas
Las atelectasias, alteración de la relación (V/Q) y la hipoventilación, resultarán en hipoxemia e hipercapnia. Los gases arteriales muestran una acidosis metabólica respiratoria que causa vasocontricción pulmonar, resultando en la alteración de la integridad endotelial y epitelial con pérdida de exudado de proteínas (como se muestra en la imagen anterior) y formación de membranas hialinas.
La deficiencia de antioxidantes y la lesión causada por la previa liberación de radicales libres como ROS, empeoran la lesión pulmonar. Las membranas hialinas que recubren los alvéolos pueden formarse aproximadamente media hora después del nacimiento, en los RNPT más grandes, el epitelio comienza a sanar a las 36-72 horas después del nacimiento, incluso puede comienzar la síntesis de surfactante endógeno.
Bibliografía
1. Christ LA, Munson DA. Disorders of the Respiratory System, Pediatrics, [Published on 06/06/2015] [Last modified 06/06/2015] https://clinicalgate.com/disorders-of-the-respiratory-system/ [revisado noviembre 2017]
2. Pramanik AK. Respiratory Distress Syndrome, Pediatrics: Cardiac Disease and Critical Care Medicine, [Updated: Jan 16, 2015] https://emedicine.medscape.com/article/976034-overview [revisado noviembre 2017]
3. iKNOLedge. Respiratory Disorders of the Newborn, Pulmolory and Respiratory, [Published on 01/06/2015, Last modified 01/06/2015] Disponible en: https://clinicalgate.com/respiratory-disorders-of-the-newborn/ [revisado noviembre 2017]
4. Echaide M, Autilio C, Arroyo R, Perez-Gil J. Restoring pulmonary surfactant membranes and films at the respiratory Surface, Biochimica et Biophysica Acta (BBA) – Biomembranes, 2017; Volume 1859, Issue 9, Part B: Pages 1725–1739
5. Edwards MO, Kotecha SJ, Kotecha S. Respiratory distress of the term newborn infant, Paediatr Respir Rev. 2013 Mar;14(1):29-36; quiz 36-7.
6. Kamath BD, Macguire ER, McClure EM, Goldenberg RL, Jobe AH. Neonatal mortality from respiratory distress syndrome: lessons for low-resource countries, Pediatrics. 2011 Jun;127(6):1139-46.
7. Speer CP. Neonatal respiratory distress syndrome: an inflammatory disease?, Neonatology. 2011;99(4):316-9.
8. Matthay MA, Zemans RL. The acute respiratory distress syndrome: pathogenesis and treatment, Annu Rev Pathol, 2011;6:147-63.
9. Matthay MA, Ware LB y Zimmerman GA. The acute respiratory distress síndrome, J Clin Invest. 2012; 122(8): 2731–2740.
Aviso de privacidad
Diseño: A. Victoria Pérez