__
__
__
EDUCACIÓN MÉDICA
ENFERMEDAD VASCULOCEREBRAL HEMORRÁGICA
FISIOPATOLOGÍA
La enfermedad cerebrovascular hemorrágica representa aproximadamente el 13% de los casos de enfermedad cerebrovascular; la hemorragia ocurre como resultado de una ruptura de un vaso debilitado; las dos etiologías de vasos debilitados generalmente causantes de derrame son: aneurismas y malformaciones arteriovenosas. La sangre que se acumula en una hemorragia comprime el tejido cerebral circundante aumentando la presión intracraneana; puede localizarse de forma intracerebral o parenquimatosa y subaracnoidea.
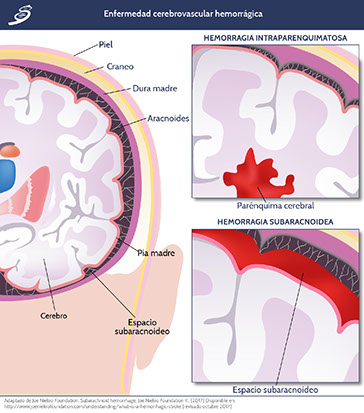
Hemorragia intracraneal parenquimatosa
La hemorragia intracraneal (HIC) consta de tres fases distintas: (1) hemorragia inicial, (2) expansión del hematoma y (3) edema peri-hematoma. La expansión del hematoma es progresivo, e implica un aumento de la presión intracraneal (PIC) que altera la integridad del tejido local y la barrera hematoencefálica (BHE). Además, el flujo de salida venoso obstruido induce la liberación de tromboplastina tisular, lo que produce coagulopatía local. En más de un tercio de los pacientes, la expansión del hematoma está asociada con hiperglucemia, hipertensión y anticoagulación. Después de la expansión, el edema cerebral se forma alrededor del hematoma, secundario a la inflamación y la alteración de la BHE. El edema peri-hematoma es causante del deterioro neurológico. En hasta 40% de los casos de HIC, ésta se extiende a los ventrículos cerebrales causando hemorragia intraventricular (HIV), asociado con la hidrocefalia obstructiva aguda. La HIC y el edema causarán compresión del tejido cerebral adyacente, conduciendo también a una disfunción neurológica e incluso, potencial herniación fatal.
Inflamación
Normalmente, la microglía ejerce un papel neuroprotector en el cerebro; inmediatamente después de una HIC, la microglía se activa como un sistema fagocitario primario que promueve la “limpieza” del hematoma. Sin embargo, los productos de la degradación del hematoma pueden activar las células de la microglía de manera excesiva, iniciando cascadas de vías de señalización inflamatoria y liberación de citocinas, quimiocinas, radicales libres y otras sustancias citotóxicas que aumentan el grado de lesión cerebral. Las citocinas inflamatorias incluyen principalmente: interleucina-1 (IL-1), IL-6 y factor de necrosis tumoral-α (TNF-α). Después, durante los días 1-3 luego de la HIC, existe mayor activación del factor nuclear κB (NF-κB) y producción de IL-1β, IL-6 y metaloproteinasa-9.
Los componentes de los hematomas, principalmente eritrocitos y sus productos de degradación como grupos hemo e iones de hierro, así como productos de la trombina, promueven la inflamación (Como se observa en la siguiente imagen). Los receptores de tipo Toll (TLR) no sólo reconocen las señales moleculares de diferentes patógenos, sino que también reciben señales de muerte celular y activan las respuestas inmunes, lo que lleva al daño tisular. Es así como después de una HIC, los TLR son activados, jugando un papel clave en la inmunidad innata y las respuestas inflamatorias. Los TLR2 y TLR4 se expresan en varias células del sistema nervioso central, incluyendo células de la microglía, astrocitos, neuronas y células endoteliales; las vías de señalización de dichos receptores son cruciales para la inflamación en HIC.

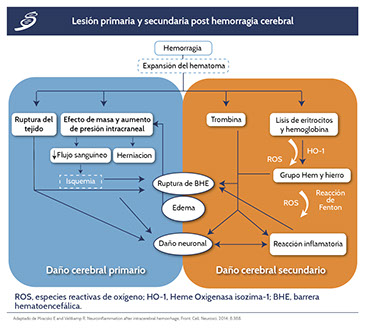
Lesion primaria
La lesión cerebral después de una HIC se divide ampliamente en primaria y secundaria; después de la ruptura repentina de los vasos sanguíneos cerebrales, el hematoma comprime el parénquima cerebral circundante, lo que lleva a un aumento agudo de la PIC, que es, como tal, la causa de la lesión cerebral primaria.
Lesion secundaria
La lesión cerebral secundaria está mediada por el grado de lesión primaria (por ejemplo, efecto de masa, PIC y estrés mecánico), así como la respuesta fisiológica al hematoma y los productos de degradación del hematoma.
Lesiones concomitantes intraparenquimatosas
Dentro de hemorragias intraparenquimatosas se han observado lesiones concomitantes sugerentes de isquemia aguda, estas lesiones se han descrito en 13-39% de las etiologías de hemorragia, tales como hipertensiva y angiopatía amiloide. De acuerdo con estudios clínicos, se ha observado que la aparición paradójica de los procesos hemorrágicos e isquémicos es más común en pacientes con alguno de los siguientes antecedentes: infarto cerebral isquémico, cambios en sustancia blanca, mayor carga amiloide, disminución abrupta de la presión arterial en las últimas 24 hrs, o creaniectomía previa; sin embargo, se desconocen los mecanismos exactos para el desarrollo de estas lesiones.
Hemorragia subaracnoidea
La hemorragia subaracnoidea (HSA) representa aproximadamente el 5% de las etiologías de la enfermedad cerebrovascular. La HSA causa un aumento de la PIC que altera la autorregulación cerebral, esto puede ocurrir en conjunto con vasoconstricción aguda y agregación plaquetaria en la microvasculatura, disminuyendo así la perfusión microvascular y, secundariamente, reducción en el flujo sanguíneo cerebral desencadenando isquemia.
Depolarización cortical
La progresión del edema perihematoma (EPH) es una complicación frecuente de HIC espontánea y se asocia con deterioro neurológico y mal pronóstico. Además, existe evidencia de que en pacientes con HIC espontanea, ocurre difusión de la despolarización (DD) principalmente en pacientes con hematomas de gran tamaño. En un estudio de microdiálisis cerebral en pacientes con HSA, se encontró una asociación entre la carga de la hemorragia inicial y concentraciones de potasio (K+) extracelular cerebral mayores. Los posibles mecanismos de aumento de los niveles de K+ en el líquido extracelular incluyen eritrocitólisis, ruptura de la BHE y ruptura de membrana inespecífica por lesión del parénquima.
La lesión cerebral temprana se informó como una causa primaria de mortalidad en pacientes con SAH, y se reconoció que muchos mecanismos patológicos importantes se iniciaron en cuestión de minutos después del aneurisma.
En humanos, el vasoespasmo cerebral generalmente ocurre el día 3 después de la HSA; existen dos picos en el día 6-8 y dura 2-3 semanas. Se considera que la isquemia cerebral retardada es inducida por vasoespasmo cerebral debido a que se encontró una fuerte asociación entre el vasoespasmo confirmado radiológicamente y los signos clínicos de retraso cerebral isquemia.
Se han demostrado muchos mediadores patofisiológicos en vasoespasmo cerebral, tales como (1) la disfunción de la vía de óxido nítrico (NO) -oxidasa sintética (NOS), (2) endotelina-1, (3) estrés oxidativo secundario a la hemoglobina ferrosa liberada del coágulo subaracnoideo, (4) vías inflamatorias, (5) ruptura de la BHE por apoptosis endotelial o trombina, (6) excitotoxicidad y patología de la membrana de los canales de Calcio (Ca++).
En una ruptura aneurismática se cree que existe un bloqueo de la circulación intracraneal por un pico inmediato de la PIC (se encuentra a la par de la presión arterial media al minuto del ictus) contribuyendo a una lesión isquémica global severa con pérdida de la autorregulación y disminución así del FSC; luego, la PIC cae durante varios minutos hasta una basal más baja, sin dejar de ser mayor a la normal. El estado hipóxico que se mantiene después de estos cambios en las presiones de perfusión culmina en inicio de cascadas inflamatorias, tales como: enfermedad cerebrovascular isquémica con fallas en la producción de adenosin trifosfato (ATP) neuronal y de células de la glia, llevando al edema citotóxico, apoptosis celular, disrupción en la barrera hematoencefálica y edema vasogénico. Las alteraciones previas forman parte de las variables patofisiológicas durante el periodo de lesión cerebral temprana (dentro de las primeras 72h).

Vasoespasmo
Se ha demostrado una disminución de la capacidad vasodilatadora en la microcirculación cerebral en pacientes que sufrieron HSA, se han descrito observaciones de las pequeñas arterias intraparenquimatosas y piales que sugieren la presencia de espasmo microvascular, especialmente un estrechamiento luminal máximo entre los días 3 y 7.
Bibliografía
AHA/ASA. Hemorrhagic Strokes (Bleeds), American Stroke Asociation, [Updated:Apr 26,2017] Disponible en: http://www.strokeassociation.org/STROKEORG/AboutStroke/TypesofStroke/HemorrhagicBleeds/Hemorrhagic-Strokes-Bleeds_UCM_310940_Article.jsp [revisado octubre 2017]
Arsava EM, Kayim-Yildiz O, Oguz KK, Akpinar E, Topcuoglu MA. Elevated admission blood pressure and acute ischemic lesions in spontaneous intracerebral hemorrhage, J Stroke Cerebrovasc Dis, 2013; 22(3):250-4.
Liebeskind DS. Hemorrhagic Stroke, Medscape Emergency Medicine, [Updated: Jan 23, 2017], disponible en: https://emedicine.medscape.com/article/1916662-overview#a5 [revisado octubre 2017]
Magistris F, Bazak S y Martín J. Intracerebral Hemorrhage: Pathophysiology, Diagnosis and Management, Clinical Review, 2013; Volume 10 No. 1: 15-22.
Chen S, Yang Q, Chen G, Zhang JH. An update on inflammation in the acute phase of intracerebral hemorrhage, Transl Stroke Res, 2015; 6(1):4-8.
Fujii M, Yan J, Rolland WB, Soejima Y, Caner B, Zhang JH. Early brain injury, an evolving frontier in subarachnoid hemorrhage research, Transl Stroke Res. 2013 Aug;4(4):432-46.
Budohoski KP, Guilfoyle M, Helmy A, Huuskonen T, Czosnyka M, Kirollos R. The pathophysiology and treatment of delayed cerebral ischaemia following subarachnoid haemorrhage, J Neurol Neurosurg Psychiatry, 2014; 85(12):1343-53.
Helbok R, Schiefecker AJ, Friberg C, Beer R, Kofler M, Rhomberg P, et al. Spreading depolarizations in patients with spontaneous intracerebral hemorrhage: Association with perihematomal edema progression, J Cereb Blood Flow Metab. 2017; 37(5):1871-1882.
Arsava EM, Kayim-Yildiz O, Oguz KK, Akpinar E, Topcuoglu MA. Elevated admission blood pressure and acute ischemic lesions in spontaneous intracerebral hemorrhage, J Stroke Cerebrovasc Dis, 2013; 22(3):250-4.
Joe Niekro Foundation. Subarachnoid hemorrhage, Joe Niekro Foundation ®, [2017] Disponible en: http://www.joeniekrofoundation.com/understanding/what-is-a-hemorrhagic-stroke [revisado octubre 2017]
Mracsko E and Veltkamp R. Neuroinflammation after intracerebral hemorrhage, Front. Cell. Neurosci, 2014; 8:388.
Starr C. Spreading depolarizations trigger early brain injury after subarachnoid hemorrhage, researchers find, Medicalxpress, [September 6, 2017] Disponible en: https://medicalxpress.com/news/2017-09-depolarizations-trigger-early-brain-injury.html [revisado octubre 2017]
Pradilla G, Chaichana KL, Hoang S, Huang J, Tamargo RJ. Inflammation and Cerebral Vasospasm After Subarachnoid Hemorrhage, 2010; Volume 21, Issue 2, Pages 365–379.
Aviso de privacidad
Diseño: A. Victoria Pérez