__
__
__
EDUCACIÓN MÉDICA
EPILEPSIA
Fisiopatología
La epilepsia es un trastorno neurológico crónico que se caracteriza por la repetición de dos o más convulsiones no provocadas, cuya manifestación clínica consiste en episodios anormales repentinos y transitorios de origen motor, sensorial, autónomo o psíquico; resultado de descargas eléctricas excesivas en un grupo de neuronas en el cerebro con resultados de comportamiento dependiendo de las regiones cerebrales donde se produzca el disparo sincrónico de un grupo de células neuronales.
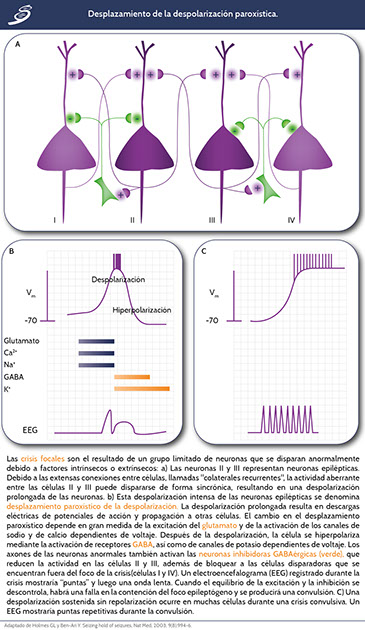
El desplazamiento de la despolarización paroxística (PDS por paroxysmal depolarization shift) es el fenómeno celular fisiopatológico que subyace a todos los tipos de crisis epilépticas y las alteraciones electroencefalográficas (EEG) de tipo epileptiformes ("en puntas"). Los PDS son eventos celulares en los que los potenciales de acción rápidamente repetitivos no son seguidos por un periodo refractario habitual, generando así una despolarización prolongada de la membrana (que es más prolongada de lo que suele ocurrir en respuesta a los potenciales postsinápticos excitatorios normales).
Potenciales de membrana neuronal
Las neuronas están compuestas por el cuerpo celular (soma) y procesos citoplasmáticos (axones y dendritas) que son extensión del soma. Las membranas celulares de las neuronas tienen varios canales iónicos que incluyen canales rápidos de sodio y potasio, así como canales de sodio y potasio regulados por voltaje que permiten el paso de iones dependiendo de su gradiente electroquímico. El potencial equilibrio para un ion es el potencial de membrana en el cual no hay movimiento neto de ese ion a través de la membrana celular; el potencial de membrana en reposo de una neurona es típicamente de -70 mV, siendo el interior de la neurona negativo en relación con el medio extracelular. El potencial de membrana de reposo se determina por el movimiento de iones de potasio, sodio y cloruro a lo largo de su gradiente electroquímico a través de la membrana celular.
Se genera un potencial de acción cuando la negatividad del potencial de membrana en reposo disminuye hasta un punto crítico (alrededor de -40 mV para las neuronas). Los canales de sodio controlados por voltaje juegan un papel importante en la generación y propagación del potencial de acción al permitir que el sodio entre en el soma, una vez generado el potencial de acción, una onda de despolarización de alta amplitud (normalmente menos de 2 ms) viaja a través de los procesos neuronales y alcanza la sinapsis.
Cada sinapsis tiene un terminal presináptico que contiene neurotransmisores en vesículas y un terminal postsináptico que contiene receptores para los neurotransmisores. Los neurotransmisores se liberan de la terminal presináptica cuando un potencial de acción provoca un cambio suficiente en el voltaje (despolarización) en el terminal presináptico para activar los canales de calcio controlados por voltaje permitiendo que el calcio entre en el terminal presináptico.
La unión de neurotransmisores a los receptores en el terminal postsináptico activa los canales iónicos, permitiendo el paso de iones que conducen a cambios locales en el potencial de membrana, llamados potenciales postsinápticos (PSPs) que son potenciales no propagados de pequeña amplitud que duran 10-100 ms. Los PSP pueden ser excitatorios (potenciales postsinápticos excitatorios [EPSP]) o inhibitorios (potenciales postsinápticos inhibitorios [IPSP]) dependiendo del tipo de canal iónico activado y del gradiente electroquímico para iones que pueden pasar a través del canal. Los EPSPs generalmente resultan de un flujo hacia dentro de iones positivos como sodio o calcio y provocan despolarización (excitación), disminuyendo así el umbral para desencadenar un potencial de acción en la terminal postsináptica. Por otro lado, los IPSPs son el resultado de un flujo interno de iones negativos (por ejemplo, cloruro) o flujo hacia fuera de iones positivos (por ejemplo, potasio) y causan hiperpolarización (inhibición), aumentando así el umbral para desencadenar un potencial de acción en el terminal postsináptico.
Crisis epilépticas
Los estados patológicos neuronales en los cuales se genera PDS, pueden basarse en propiedades neuronales intrínsecas:
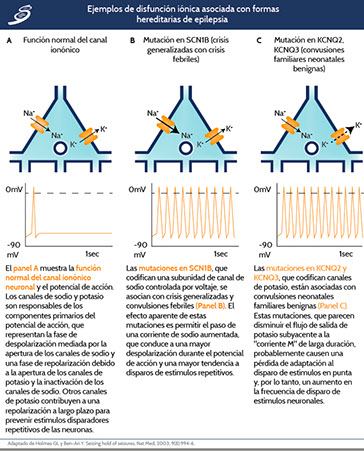
•Disfunción de los canales iónicos en ciertas canalopatías determinadas genéticamente, se muestra un ejemplo gráfico de las canalopatías en la siguiente imagen.
•Concentraciones inhibitorias inadecuadas de neurotransmisores (principalmente de ácido gamma-aminobutírico o GABA).
•Exposición a concentraciones elevadas de neurotransmisores exitatorios (el principal glutamato) o aminoácidos exitatorios externos.
Sin embargo, la PDS debe ser generada por un grupo de neuronas simultáneamente para conducir a la naturaleza episódica de las convulsiones.
Crisis parciales
Las crisis parciales o de inicio focal se observan en el EEG como ondas agudas o puntas epileptiformes. La correlación neurofisiológica celular es la de una descarga epiléptica focal intersticial de un solo grupo de neuronas corticales con PDS.
El PDS se caracteriza por una depolarización prolongada dependiente del calcio que da lugar a múltiples potenciales de acción mediados por sodio durante la fase de despolarización, seguida por una prominente hiperpolarización posterior, que es un potencial de membrana hiperpolarizado más allá del potencial de reposo basal. Los canales de potasio dependientes del calcio median, en mayor parte, la fase posterior a la hiperpolarización. Cuando múltiples neuronas disparan PDSs de una manera sincrónica, se muestran puntas intersticiales.
Los siguientes mecanismos pueden coexistir en diferentes combinaciones para causar convulsiones de inicio focal:
- Disminución de la inhibición
- Activación defectuosa de las neuronas GABA
- Aumento de la activación
Disminución de la inhibición
El principal causante de la disminución en la eficacia de la inhibición es secundario al neurotransmisor GABA. Existen dos circunstancias principales en las que la inhibición GABAérgica puede degradarse rápidamente: poca liberación de GABA y demasiada liberación de GABA. Una manera más sencilla de expresar estos mecanismos presinápticos es un desajuste ictogénico en la liberación de GABA.
Es probable que el desajuste temporal o espacial de la liberación de GABA pueda provocar convulsiones.
Los mecanismos que conducen a una disminución de la inhibición incluyen los siguientes:
- Inhibición de GABA-A defectuosa
- Inhibición de GABA-B defectuosa
- Activación defectuosa de las neuronas GABA
- Defecto intracelular de calcio como buffer
Activación defectuosa de las neuronas GABA
Las neuronas GABA se activan por medio de señales compensadoras y de retroalimentación de neuronas excitadoras. Estos dos tipos de inhibición en la red neuronal son definidas con base en el tiempo de activación de las neuronas GABAérgicas en relación con la principal salida neuronal de la red, como se observa con la célula hipocámpica piramidal CA1.
Aumento de la activación
Los mecanismos que conducen al aumento de la excitación son los siguientes:
- Aumento de la activación de los receptores NMDA
- Aumento de la sincronía entre las neuronas debido a una transmisión efáptica (transmisión de un impulso nervioso de una fibra a otra a través de las membranas, no de sinapsis)
- Aumento de la sincronía y/o activación debido a la recurrencia de estímulos colaterales excitatorios
Crisis generalizadas
El mejor ejemplo es la interacción tálamo-cortical que puede servir de base a las crisis de ausencia típicas. El circuito talamocortical tiene ritmos oscilatorios normales, con peroiodos de aumentos relativos de la excitación y aumentos relativos de la inhibición; es el que genera las oscilaciones observadas en los patrones de sueño. Los circuitos talamocorticales incluyen las neuronas piramidales del neocortex, las neuronas retransmisoras talámicas y las neuronas del núcleo reticular del tálamo (imagen con inciso A).

Los ritmos talamocorticales alterados pueden dar lugar a convulsiones generalizadas primarias. Las neuronas retransmisoras talámicas reciben entradas ascendentes de la médula espinal y proyectan a las neuronas neocorticales piramidales. Las vías colinérgicas del prosencéfalo y las vías ascendentes serotoninérgicas, noradrenérgicas y colinérgicas del tronco encefálico regulan de manera prominente este circuito.
Las neuronas retransmisoras talámicas pueden tener oscilaciones en el potencial de membrana en reposo, el cual aumenta la probabilidad de activación sincrónica de las neuronas piramidales neocorticales durante la despolarización, lo que reduce significativamente la probabilidad de activación neocortical durante la hiperpolarización relativa. La clave de estas oscilaciones es el canal de calcio transitorio de bajo umbral, también conocido como corriente T-calcio.
En la Figura anterior inciso B se ejemplifica en EEG cómo, durante la vigilia, la corteza es activada por el tálamo en modo tónico que permite el procesamiento de entradas sensoriales externas, esto da una apariencia desincronizada en el EEG. Durante el sueño No-REM, la corteza se activa en modo ráfaga, lo que resulta en el EEG como los husos rítmicos del sueño. Durante una crisis de ausencia, el circuito tálamo-cortical normal se vuelve disfuncional, permitiendo la activación en modo ráfaga de la corteza que ocurre durante la vigilia, lo que da como resultado un EEG de descargas rítmicas generalizadas punta-onda e interrumpe la respuesta a estímulos externos.
Referencias
Cerri C, Caleo M y Bozzi Y. Chemokines as new inflammatory players in the pathogenesis of epilepsy, Epilepsy Res. 2017; 136:77-83.
Thomas RH. Seizures and Epilepsy: Pathophysiology and Principles of Diagnosis, Epilepsy Board Review Manual, Copyright 2012 by Turner White Communications Inc., Wayne, PA. All rights reserved, Volume I, Part I, 1-26.
Bandyopadhyay S, Koubeissi MZ y Azar NJ. Physiologic Basis of EEG and Epilepsy, Epilepsy Board Review, 2017; 1-11.
Trevelyan AJ, Muldoon SF, Merricks EM, Racca C y Staley KJ. The role of inhibition in epileptic networks, J Clin Neurophysiol, 2015; 32(3):227-34.
Ko DY. Pathophysiology, Epilepsy and Seizures, [Updated: Jul 12, 2016] Disponible en: http://emedicine.medscape.com/article/1184846-overview#a3 [Revisado agosto 2017]
Holmes GL y Ben-Ari Y. Seizing hold of seizures, Nat Med, 2003; 9(8):994-6.
Chang BS y Lowenstein DH. Epilepsy, N Engl J Med, 2003; 349(13):1257-66.
Aviso de privacidad
Diseño: A. Victoria Pérez