__
__
__
EDUCACIÓN MÉDICA
DEPRESIÓN MAYOR
FISIOPATOLOGÍA NEUROBIOQUÍMICA
La depresión es un trastorno mental crónico que causa cambios en el estado de ánimo, los pensamientos, el comportamiento y la salud física. De acuerdo al Manual Diagnóstico y Estadístico de los Trastornos Mentales, 5ª edición (DSM-V), el rasgo distintivo del trastorno depresivo mayor es la aparición del estado de ánimo deprimido (disforia) y la pérdida de interés en actividades que eran bastante placenteras en el pasado (anhedonia) durante al menos dos semanas.
Es un trastorno heterogéneo con un curso muy variable, una respuesta inconsistente al tratamiento en muchos de los casos y ningún mecanismo establecido; sin embargo, en investigaciones resientes se han logrado describir mecanismos que se ven inmiscuidos en la depresión, como los que se mencionan a continuación.
Vulnerabilidad genética e interacción ambiental
El surgimiento de la depresión se debe a una compleja interacción entre condiciones genéticas y ambientales que alteran la respuesta individual a situaciones de vida estresantes. Ningún polimorfismo genético parece ser responsable de causar depresión, se ha sugerido que los factores genéticos hacen que ciertos individuos sean susceptibles a la depresión al aumentar su vulnerabilidad a factores ambientales estresantes durante años; es la variación alélica en la región promotora del gen que codifica el transportador de serotonina (5- HTT). La región promotora del gen 5-HTT (5-HTTLPR) contiene un polimorfismo funcional que da como resultado una variante larga (L)/corta (S) en la región promotora corriente, arriba del sitio de inicio de la transcripción. El alelo corto de 5-HTT tiene una baja actividad y se ha demostrado que pone a los portadores en un mayor riesgo de desarrollar depresión en respuesta a eventos estresantes de la vida. Este alelo también se ha relacionado con peores resultados después de los tratamientos farmacológicos y no farmacológicos antidepresivos. La enzima limitante de la velocidad en la biosíntesis de serotonina, triptófano hidroxilasa (TPH), está codificada por dos genes distintos: Tph1 y Tph2, y se ha propuesto que desempeña un papel en la patogénesis de los trastornos depresivos y el suicidio. Los polimorfismos de un solo nucleótido (SNP) en el gen Tph2 se han relacionado con una mayor incidencia de depresión y han completado intentos de suicidio.
Además, se cree que el gen Tph1, que se expresa predominantemente en la glándula pineal, influye en el riesgo suicida al alterar la síntesis de la melatonina, hormona responsable de la regulación del ritmo circadiano que da como resultado un aumento en el riesgo suicida. Un polimorfismo funcional, produciendo una sustitución de valina a metionina en el codón 66 (Val66Met) en la región pro-BDNF, se ha identificado en el gen BDNF (factor neurotrófico derivado del cerebro, brain-derived neurotrophic factor) exhibiendo un efecto perjudicial sobre el tráfico intracelular y la secreción dependiente de la actividad, e influenciando la función del hipocampo, memoria episódica y morfología cerebral. Las personas sanas con la variante BDNF en el alelo Met muestran una baja estabilidad emocional y un volumen menor de hipocampo. Los estudios también sugieren que existe una interacción compleja entre los polimorfismos en los genes que codifican BDNF y 5- HTT para producir un fenotipo deprimido.
La vulnerabilidad ambiental tiene una gran influencia en el desarrollo neuronal, así como la neuroplasticidad, pero no se abordará en este tema, en cambio hablaremos de las deficiencias neurobiológicas sin dejar de lado que las circunstancias ambientales (la vida diaria) tiene un papel desencadenante en la mayoría (no todos) de los casos.
Deficiencia de monoamina
Los sistemas noradrenérgico y serotoninérgico se expanden en casi todo el cerebro, lo que sugiere que es un sistema capaz de modular muchas áreas cerebrales que involucran sentimiento, pensamiento y comportamiento. Posterior al uso de los primeros antidepresivos se produjo el descubrimiento de una importante teoría de la depresión conocida como la hipótesis de la deficiencia de monoaminas.
Una forma de poder corroborar su relación, es por medio de estudios tomográficos de emisión de positrones utilizando un ligando para monoamina oxidasa cerebral: en estudios donde se ha utilizado este procedimiento, se ha logrado demostrar un aumento del 30% de la enzima en subgrupos de pacientes con depresión. Un estudio que mide las diferencias en metabolitos de monoaminas entre la vena yugular interna y la arteria braquial, el cerebro mostró una menor producción de los metabolitos de la norepinefrina en pacientes con depresión que en los controles. La hipótesis de la deficiencia de monoaminas continúa estimulando la investigación cuando se abre una nueva ventana técnica en el cerebro.
Otra manera de realizar el estudio de monoaminas y su efecto en la depresión es mediante los efectos posteriores de la neurotransmisión de monoaminas. El receptor de serotonina-1B se localiza presinápticamente y regula la liberación de serotonina por inhibición de retroalimentación. El receptor de serotonina-1A se localiza tanto presinápticamente como postsinápticamente para regular la función de la serotonina. El receptor puede evaluarse en pacientes con depresión mediante la administración de agonistas específicos y midiendo las respuestas neuroendocrinas específicas, como la elevación del nivel de prolactina. Los resultados de estas mediciones sugieren que la sensibilidad de este receptor se reduce en pacientes con depresión. Por otro lado, el receptor α2-noradrenérgico, que generalmente es presináptico, modula la liberación de norepinefrina por inhibición de retroalimentación, describiéndose una mayor sensibilidad del receptor en pacientes con depresión, concordando con la liberación reducida de norepinefrina. Las mediciones directas de la neurotransmisión de monoaminas exploraron los efectos posteriores de la neurotransmisión de monoaminas, así como el papel del receptor de serotonina-1A que se localiza tanto presinápticamente como postsinápticamente para regular la función de la serotonina, pudiéndose observar en la siguiente imagen.
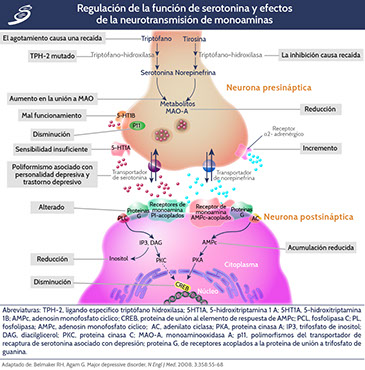
La hipótesis de la depresión con monoaminas postula una deficiencia en la neurotransmisión de serotonina o norepinefrina en el cerebro. La neurotransmisión monoaminérgica está mediada por la serotonina (5-hidroxitriptamina 1A [5-HT1A] y 5-hidroxitriptamina 1B [5-HT1B]) o la norepinefrina (noradrenalina) liberada por las neuronas presinápticas (neurona serotoninérgica, que se muestra en el lado izquierdo y neurona noradrenérgica, mostrado en el lado derecho [condensado virtualmente en la imagen anterior]). La serotonina se sintetiza a partir de triptófano, con el primer paso en la ruta sintética catalizada por la enzima triptófano hidroxilasa; la norepinefrina se sintetiza a partir de la tirosina, con el primer paso catalizado por la enzima tirosina hidroxilasa. Ambos transmisores de monoaminas se almacenan en vesículas en la neurona presináptica y se liberan en la hendidura sináptica, afectando de ese modo a las neuronas presináptica y postsináptica. La cesación de la acción sináptica de los neurotransmisores ocurre por medio de la recaptura a través de los transportadores específicos de serotonina y norepinefrina y el control de la liberación mediante los autoreceptores presinápticos de 5-HT1A y 5-HT1B para la serotonina, y los autoreceptores α2-noradrenérgicos para norepinefrina. La monoaminooxidasa A (MAO-A) cataboliza las monoaminas presinápticamente y por lo tanto regula indirectamente el contenido vesicular. La proteína p11 (proteína de unión a calcio S100 A10), que interactúa con los receptores 5-HT1B, aumenta su función. Postsinápticamente, tanto la serotonina como la norepinefrina se unen a dos tipos de receptores acoplados a la proteína de unión a trifosfato de guanina (proteína G): receptores acoplados a AMP cíclico (AMPc) que activan la adenilato ciclasa (AC) para generar AMPc y fosfatidilinositol (PI) como receptores acoplados, que activan la fosfolipasa C (PLC). PLC genera trifosfato de inositol (IP3) y diacilglicerol (DAG); el AMPc activa la proteína cinasa A (PKA) e IP3 y DAG activan la proteína cinasa C (PKC). Las dos proteínas cinasas afectan a la proteína de unión al elemento de respuesta de AMPc (CREB). Los hallazgos en pacientes con depresión que apoyan la hipótesis de deficiencia de monoamina incluyen: evidencia de recaídas a estados depresivos secundarios, a inhibición de la tirosina hidroxilasa o el agotamiento del triptófano en la dieta, aumento en la frecuencia de una mutación que afecta la forma específica del ligando específico triptófano hidroxilasa (TPH-2) que se une a la MAO-A, receptores 5-HT1A con sensibilidad insuficiente, receptores 5-HT1B con mal funcionamiento, niveles disminuidos de p11, polimorfismos del transportador de recaptura de serotonina asociado con depresión, una respuesta inadecuada de las proteínas G a señales de neurotransmisores y niveles reducidos de AMPc, inositol y CREB, todo esto en cerebros post mortem.
El papel de segundos mensajeros, compuestos orgánicos
y monoaminas en la depresión
Es concebible que los sistemas de segundo mensajero para la neurotransmisión serotoninérgica y noradrenérgica funcionen mal en la depresión, y por esta razón han sido ampliamente estudiados los sistemas de los segundos mensajeros como fosfatidilinositol y AMPc. Otro compuesto orgánico que se ha observado que tiene un papel en la depresión es el inositol. Se han encontrado niveles reducidos de inositol en estudios postmortem de cerebros de personas que han muerto por suicidio y en estudios por medio de espectroscopia de resonancia magnética de la corteza frontal en pacientes con depresión.
El AMPc también juega un papel importante en la depresión, se ha determinado en estudios postmortem que este nucleótido se encuentra reducido, por lo tanto, la función de este segundo mensajero puede perjudicar la función de los neurotransmisores, incluso sin cambios en los niveles de monoaminas o en los números de los receptores. Las proteínas G que median la señalización entre receptores y sistemas de segundo mensajero también se han investigado en pacientes con depresión, tanto en estudios postmortem como en estudios de células de sangre periférica, observándose que los sistemas se ven claramente afectados, no ha surgido una hipótesis consistente porque existen numerosas formas de proteínas G que varían en diferentes áreas del cerebro.
La proteína CREB es un factor de transcripción afectado por AMPc en la célula. Los niveles de CREB y fosfo-CREB aparecían reducidos en estudios postmortem de las cortezas cerebrales de pacientes con un trastorno depresivo mayor sin tratamiento previo, en comparación con los controles.
Muchos estudios del sistema de segundos mensajeros y factores de transcripción en la depresión se inspiraron en la creencia de que transcurren varias semanas antes de que el tratamiento con antidepresivos tenga efecto; en consecuencia, los estudios fueron diseñados para detectar cambios bioquímicos dependientes del tiempo en las células nerviosas. Sin embargo, los nuevos metanálisis sugieren que los efectos antidepresivos comienzan rápidamente, lo que respalda la clásica hipótesis de la deficiencia de monoaminas. Un punto fuerte de la teoría de las monoaminas ha sido su poder predictivo, por ello, farmacológicamente hablando, casi todos los compuestos que se han sintetizado con el propósito de inhibir la recaptura de norepinefrina o serotonina han demostrado ser antidepresivos clínicamente eficaces.
El estrés, el eje hipotalámico-hipofisario-suprarrenal
y los factores de crecimiento
El estrés es percibido por la corteza del cerebro y se transmite al hipotálamo, donde la hormona liberadora de corticotropina (CRH) se libera en los receptores de la hipófisis. Este estímulo da como resultado la secreción de corticotropina en el plasma, la estimulación de los receptores de corticotropina en la corteza suprarrenal y la liberación de cortisol en la sangre. Los receptores de cortisol hipotalámico responden disminuyendo la producción de CRH para mantener la homeostasis.
Existe considerable evidencia de que el cortisol y su factor de liberación central, CRH, están involucrados en la depresión. Los pacientes con depresión pueden tener niveles elevados de cortisol en el plasma, niveles elevados de CRH en el líquido cefalorraquídeo y niveles elevados de ARN mensajero, CRH y proteínas en las regiones del sistema límbico. En estudios que usan dexametasona para evaluar la sensibilidad del hipotálamo a señales de retroalimentación para el cierre de la liberación de CRH, la respuesta normal de supresión de cortisol está ausente en aproximadamente la mitad de los pacientes con depresión más severa, e incluso se ha descrito que la remisión clínica inducida por antidepresivos se acompaña de la reversión de algunas de estas anomalías.
El aumento de los niveles de monoaminas en la sinapsis afecta el eje hipotálamo-hipófisis-adrenal y revierte algunos de los efectos a largo plazo del estrés, es por ello que se cree en la posibilidad de que los antidepresivos alivien la depresión al reducir el estrés secundario causado por un estado de ánimo dolorosamente desanimado en lugar de elevar directamente el estado de ánimo.
Una de las principales responsabilidades de la teoría hipotalámica-pituitaria-suprarrenal del eje de la depresión es la dificultad de lograr definir la relación existente entre el estrés y la depresión. Algunos pacientes tienen un único episodio depresivo de por vida, mientras que una mayor proporción tiene un curso recurrente o incluso crónico. Varios tipos de estrés agudo, trauma en la primera infancia o problemas psicosociales a largo plazo pueden estar involucrados y pueden llevar a respuestas diferentes del sistema de estrés. El estrés puede ser causante en algunos casos y secundario al estado de ánimo deprimido en otros. En la siguiente imagen se puede observar el mecanismo del eje hipotálamo-hipófisis-adrenal y su papel en la depresión.
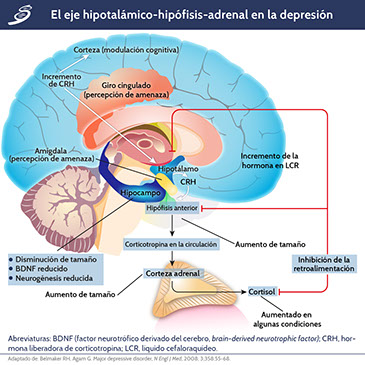
La imagen previa muestra la hipótesis sobre el eje hipotálamo-hipófisis-adrenal; las anomalías en la respuesta del cortisol al estrés pueden ser la base de la depresión. Las flechas negras muestran que, en respuesta al estrés, que es percibido por la corteza cerebral y la amígdala y se transmite al hipotálamo, se libera CRH, lo que induce a la hipófisis anterior a secretar corticotropina en el torrente sanguíneo. La corticotropina estimula la corteza suprarrenal para secretar la hormona glucocorticoide cortisol. Las líneas rojas muestran que el cortisol, a su vez, induce la inhibición de la retroalimentación en el hipotálamo y la hipófisis, suprimiendo la producción de CRH y corticotropina, respectivamente. El tamaño del hipocampo y el número de neuronas y neuroglia están disminuidos, posiblemente reflejando reducción de la neurogénesis debido a niveles elevados de cortisol o debido a la reducción de BDNF.
La enseñanza clásica consiste en que las neuronas no se dividen en el cerebro de un mamífero adulto, pero los estudios han demostrado que la neurogénesis ocurre en varias áreas del cerebro, especialmente en el hipocampo. Los niveles elevados de glucocorticoides por el estrés pueden reducir la neurogénesis, y esto se ha sugerido como un mecanismo para la disminución del tamaño del hipocampo del cerebro en muchos pacientes con depresión y que se ha corroborado con imágenes de resonancia magnética.
La hipótesis neurotrófica de la depresión
Los factores de riesgo de episodios depresivos cambian durante el curso de la enfermedad. El primer episodio depresivo suele ser "reactivo", es decir, desencadenado por factores estresantes psicosociales importantes, mientras que los episodios posteriores se vuelven cada vez más "endógenos", es decir, desencadenados por factores estresantes menores o que ocurren espontáneamente. Existe evidencia consistente de que la pérdida de volumen del hipocampo y otras regiones cerebrales está relacionada con la duración de la depresión, lo que sugiere que la depresión no tratada conduce a la pérdida de volumen del hipocampo, lo que posiblemente incrementa la sensibilidad al estrés y aumenta el riesgo de recurrencia. La neurotoxicidad de los glucocorticoides, la toxicidad glutamatérgica, la disminución de los factores neurotróficos y la disminución de la neurogénesis se han propuesto como posibles mecanismos que explican la pérdida de volumen cerebral en la depresión. El BDNF ha atraído un interés considerable. Específicamente, los estudios preclínicos han mostrado correlaciones entre conductas depresivas inducidas por estrés y disminuciones en los niveles de BDNF en el hipocampo, así como una expresión mejorada de BDNF después del tratamiento con antidepresivos, es por ello que se ha establecido una conciencia del médico ante el efecto potencialmente dañino para el cerebro de la depresión y tratar a los pacientes deprimidos de la manera más temprana y efectiva posible.
Neurotransmisión glutamatérgica y GABAérgica alterada
Una serie de estudios de espectroscopia y resonancia magnética mostraron consistentemente reducciones en las concentraciones totales de ácido gamma-aminobutírico (GABA) en la corteza prefrontal y occipital en la depresión aguda.
Bibliografía
American Psychiatric Association. DSM-5: Manual diagnóstico y estadístico de los trastornos mentales, 5ed. Washington DC, American Psyquiatric publishing, 2014; p.74-75.
Belmaker RH, Agam G. Major depressive disorder, N Engl J Med, 2008; 3;358:55-68.
Hasler G. Pathophysiology of depression: do we have any solid evidence of interest to clinicians?, World Psychiatry, 2010;9:155-61.
Duman RS, Voleti B. Signaling pathways underlying the pathophysiology and treatment of depression: novel mechanisms for rapid-acting agents, Trends Neuroscience, 2012;35:47-56.
Fekadu N, Shibeshi W, Engidawork E. Major Depressive Disorder: Pathophysiology and Clinical Management, J Depress Anxiety, 2017, 6:1
Lopresti AL, Hood SD, Drummond PD. A review of lifestyle factors that contribute to important pathways associated with major depression: diet, sleep and exercise, J Affect Disord, 2013;148:12-27.
Aviso de privacidad
Diseño: A. Victoria Pérez