__
__
__
EDUCACIÓN MÉDICA
DIABETES MELLITUS TIPO 2
FISIOPATOLOGÍA
La diabetes tipo 2 (DM2) es una enfermedad caracterizada por un defecto tanto en la secreción como en la acción de la insulina, cuya interacción compleja conduce a un aumento progresivo de los niveles de glucosa plasmáticos.
ENFERMEDAD VASCULAR
Las alteraciones en la homeostasis vascular debidas a la disfunción endotelial y de las células musculares lisas son las características principales de la vasculopatía diabética que favorecen un estado proinflamatorio/trombótico que finalmente conduce a la aterotrombosis. Las complicaciones macro y microvasculares diabéticas se deben principalmente a la exposición prolongada a la hiperglucemia que se agrupa con otros factores de riesgo como hipertensión arterial, dislipidemia y susceptibilidad genética. Los efectos perjudiciales de la glucosa comienzan con niveles de glucemia por debajo del umbral para el diagnóstico de la diabetes.
El desencadenante inicial por el que las altas concentraciones de glucosa alteran la función vascular es el desequilibrio entre la biodisponibilidad de óxido nítrico (NO) y la acumulación de especies reactivas de oxígeno (ROS) que conduce a la disfunción endotelial. De hecho, la generación de anión superóxido inducida por hiperglucemia (O2-) inactiva el NO para formar peroxinitrito (ONOO-), un poderoso oxidante que penetra fácilmente a través de las membranas de fosfolípidos e induce la nitración del sustrato. La nitrosilación de proteínas entorpece la actividad de enzimas antioxidantes y de NO sintetasa endotelial (eNOS, como se observa en la siguiente imagen). Es importante destacar que la reducción de la biodisponibilidad es un fuerte predictor de los resultados cardiovasculares.

Las altas concentraciones de glucosa intracelular conducen a la activación de la proteína cinasa C (PKC) y posterior producción de ROS por nicotinamida adenina dinucleótido fosfato (NADPH) oxidasa y la proteína adaptadora p66Shc. El aumento del estrés oxidativo inactiva rápidamente el NO que conduce a la formación del ONOO- pro-oxidante, responsable de la nitrosilación de proteínas. La disponibilidad reducida de NO también se debe a la disregulación de eNOS dependiente de PKC. De hecho, la PKC desencadena una regulación al alza de enzimas que mejoran el desacoplamiento de eNOS y conduce a una mayor acumulación de radicales libres. Por otro lado, la hiperglucemia reduce la actividad de eNOS que entorpece la fosforilación activadora en Ser1177. Junto con la falta de NO, la activación de PKC inducida por la glucosa provoca una síntesis aumentada de ET-1 que favorece la vasoconstricción y la agregación plaquetaria. La acumulación de anión superóxido también desencadena la regulación positiva de los genes proinflamatorios: proteína quimiotáctica de monocitos-1 (MCP-1), molécula de adhesión de células vasculares-1 (VCAM-1) y molécula de adhesión celular intracelular-1 (ICAM-1) a través de la activación de la señalización del factor nuclear kappa-beta (NF-kB). Estos eventos conducen a la adhesión de los monocitos, movimiento y diapédesis con la formación de células espumosas en la capa subendotelial. Las citocinas inflamatorias derivadas de células espumosas mantienen la inflamación vascular, así como la proliferación de células del músculo liso, acelerando el proceso aterosclerótico. La disfunción endotelial en la diabetes también se deriva del aumento de la síntesis de tromboxano A2 (TXA2) a través de la regulación positiva de cicloxigenasa 2 (COX-2) y la inactivación de prostaciclinas (PGI2) mediante una mayor nitrosilación. Además, las ROS aumentan la síntesis del metabolito de la glucosa metilglioxal, lo que conduce a la activación de la señalización de los productos finales de la glicación avanzada/receptor para productos finales de glicosilación avanzada (AGE/RAGE) y del flujo pro-oxidante de la hexosamina y la vía de los polioles (véase imagen previa).
El entorno hiperglucémico induce una elevación crónica de los niveles de diacilglicerol en las células endoteliales con la posterior translocación de la membrana de las isoformas de PKC convencionales (α, β1, β2,) y no convencionales (δ). Una vez activado, PKC es responsable de diferentes cambios estructurales y funcionales en la vasculatura, incluyendo alteraciones en la permeabilidad celular, inflamación, angiogénesis, crecimiento celular, expansión de la matriz extracelular y apoptosis.
Tradicionalmente la vasculopatía asociada a DM se agrupa en dos subtipos principales; complicaciones microvasculares como retinopatía, nefropatía y neuropatía, y complicaciones macrovasculares aterotrombóticas como infarto al miocardio, enfermedad cerebrovascular y enfermedad arterial periférica. A continuación, se explican los mecanismos de daño de acuerdo a esta división.
ALTERACIONES MICROVASCULARES
La enfermedad microvascular tiende a ocurrir predominantemente en tejidos donde la captación de glucosa es independiente de la actividad de la insulina (por ejemplo, riñón, retina y endotelio vascular) porque estos tejidos están expuestos a niveles de glucosa que se correlacionan muy estrechamente con los niveles de glucosa en sangre.
Retinopatía diabética
Varios estudios han explorado la asociación entre la retinopatía diabética y las complicaciones macrovasculares. Como la microvasculatura retiniana comparte características embriológicas y anatómicas con la de la circulación cerebral, los investigadores han estudiado las anomalías retinianas para proporcionar pistas para comprender la fisiopatología subyacente de las diferentes enfermedades cerebrovasculares. En un estudio multinacional de la Organización Mundial de la Salud sobre la enfermedad vascular en la diabetes, la retinopatía se relacionó con la incidencia de infarto de miocardio y la muerte por enfermedad cardiovascular (ECV). También, el aumento de la incidencia de enfermedad cerebrovascular clínica se asoció con anomalías microvasculares retinianas y estrechamiento arteriolar generalizado. Además, en varios estudios se ha asociado significativamente la retinopatía diabética con incidencia de enfermedad cerebrovascular en pacientes con retinopatía proliferativa, aunque también se han reportado eventos cardiovasculares sin presencia de retinopatía.
Tradicionalmente se considera una enfermedad microvascular, pero la neurodegeneración de la retina también está involucrada. Los mecanismos fisiopatológicos desencadenados por la hiperglucemia incluyen factores genéticos y epigenéticos, aumento de la producción de radicales libres, AGE, factores inflamatorios y factor de crecimiento endotelial vascular (VEGF) (como en la siguiente imagen).
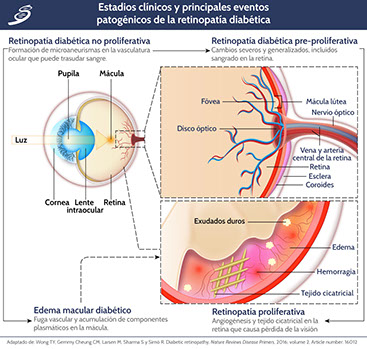
La primera etapa patológica que se puede identificar en un examen de fondo de ojo es la retinopatía diabética no proliferativa, que se puede dividir en retinopatía diabética no proliferativa leve, moderada o grave. El edema macular diabético se puede considerar un subtipo particular de retinopatía diabética no proliferativa en la que la filtración vascular afecta a la mácula. La retinopatía diabética no proliferativa pre-proliferativa o severa se caracteriza por cierres capilares y áreas no perfundidas. El hallazgo fundoscópico típico es la presencia de manchas de algodón y anormalidades microvasculares intrarretinianas. La retinopatía diabética proliferativa, en la que la hipoxia tiene un papel esencial, es la etapa final de la retinopatía diabética y su característica distintiva es el desarrollo de la neovascularización. Los hemovítreos (es decir, la hemorragia en la cavidad vítrea) y el desprendimiento de retina son complicaciones avanzadas de la retinopatía diabética proliferativa (ver imagen previa).
Nefropatía diabética
La proteinuria ocurre en 15 a 40% de los pacientes con diabetes tipo 1, mientras que oscila entre 5 y 20% de los pacientes con DM2. Según el estudio European Diabetes Prospective Complications Study, la incidencia acumulada de microalbuminuria fue del 12.6% durante 7.3 años en pacientes con DM1. Sin embargo, un estudio de seguimiento de 18 años de Dinamarca informó una tasa de prevalencia del 33% en la población de DM1. Del mismo modo, en el (UKPDS), los pacientes con DM2 mostraron una incidencia de microalbuminuria del 2.0% por año, que alcanzó hasta el 25% a 10 años después del diagnóstico. La prevalencia de nefropatía diabética fue más alta en afroamericanos, asiáticos y nativos americanos que en caucásicos.
Los mecanismos patógenos que subyacen a la nefropatía diabética implican la generación de ROS, la acumulación de AGE y la activación de moléculas de señalización intracelular como la PKC. Se ha demostrado una fuerte asociación entre la nefropatía diabética y la retinopatía, la asociación directa entre la presencia de microalbuminuria y las complicaciones macrovasculares.
Los cambios metabólicos que alteran la hemodinámica renal y promueven la inflamación y la fibrosis en la diabetes temprana incluyen hiperaminoacidemia, un promotor de la hiperfiltración, hiperperfusión glomerular e hiperglucemia. La hipertensión sistémica y la obesidad también contribuyen a la hiperfiltración glomerular a través de mecanismos, como la presión arterial sistémica y el agrandamiento glomerular. La hiperfiltración glomerular se observa en un 10% a 40% o hasta en un 75% de los pacientes con DM1, y hasta en un 40% de los pacientes con DM2; un mecanismo plausible de la hiperfiltración glomerular en la diabetes es el aumento de la reabsorción tubular proximal de la glucosa a través del cotransportador 2 sodio-glucosa, lo que disminuye el suministro distal de solutos, particularmente cloruro de sodio a la mácula densa. La disminución resultante en la retroalimentación tubuloglomerular puede dilatar la arteriola aferente para aumentar la perfusión glomerular, mientras que, al mismo tiempo, la alta producción local de angiotensina II en la arteriola eferente produce vasoconstricción. El efecto general es una alta presión intraglomerular e hiperfiltración glomerular (como en la siguiente imagen de una nefrona).

En el glomérulo suceden cambios tempranos que son críticos para el posterior desarrollo de la glomeruloesclerosis y la pérdida de la función de la nefrona. Entre estos cambios, el más importante podría ser la disfunción de los podocitos glomerulares, que son células diferenciadas terminales altamente especializadas que cubren el polo urinario de la membrana basal glomerular (GBM). Junto con las células endoteliales glomerulares, los podocitos son responsables del mantenimiento de la GBM, su barrera con carga y la forma e integridad del bucle del capilar glomerular; todas estas funciones están comprometidas en el glomérulo diabético. El medio diabético induce cambios "patopaptativos" en los podocitos, incluida la reorganización del citoesqueleto, la des-diferenciación, la apoptosis y la autofagia que se manifiesta por el ensanchamiento morfológico, la retracción y el aplanamiento (conocido como borramiento), la motilidad reducida, el aumento de la formación de uniones estrechas, una disminución en la longitud de la hendidura del diafragma, hipertrofia glomerular, desprendimiento y pérdida de los podocitos (como se observa en la siguiente imagen).
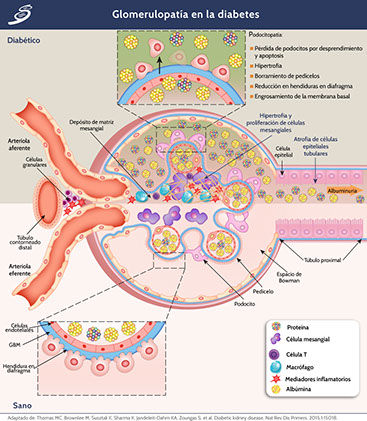
Uno de los cambios glomerulares más tempranos y característicos de la diabetes es un engrosamiento homogéneo de la GBM; éste está presente en casi todos los pacientes a los pocos años de diagnóstico. No está claro si el engrosamiento de GBM es un marcador de podocitosis o disfunción endotelial, o es un mediador de la enfermedad renal diabética progresiva. Ciertamente, los cambios en la composición, carga o arquitectura de la GBM asociados con el engrosamiento, podrían contribuir a la albuminuria. La rigidez de la GBM también podría reducir la distensibilidad de la pared pericapilar y comprometer el espacio de los subpodocitos, facilitando la lesión glomerular a través de mecanismos hemodinámicos. Las células mesangiales también están sustancialmente alteradas por la diabetes, experimentando una proliferación e hipertrofia mientras aumentan su producción de proteínas de la matriz. Estos cambios conducen a algunas de las características estructurales únicas de la glomerulopatía diabética (como en la imagen previa) incluyendo un aumento en el volumen fraccionario del glomérulo ocupado por mesangio (expansión mesangial), degeneración focal de las células mesangiales y la matriz mesangial (mesangiólisis) y, finalmente, la glomeruloesclerosis. Existe un fuerte vínculo entre la expansión de la matriz mesangial y la progresión de la enfermedad renal diabética; sin embargo, a diferencia de los podocitos, la regulación positiva del transporte de glucosa a las células mesangiales no recapitula un fenotipo diabético, lo que sugiere que la interferencia entre los podocitos, las células endoteliales e inflamatorias media la expansión de la matriz mesangial en lugar de ser un efecto directo de la exposición a la glucosa en las células mesangiales. Aunque los detalles moleculares de cómo la diabetes altera las células mesangiales no se comprenden completamente, la importancia de la expansión de la matriz mesangial en el desarrollo y la progresión de la glomeruloesclerosis asociada a la diabetes es clara. Por ejemplo, la reducción resultante en el área de la superficie capilar como resultado de la expansión del mesangio contribuye a la hipertensión glomerular, la proteinuria y la filtración glomerular reducida.
Los miofibroblastos son responsables de la deposición de la matriz que conduce a la fibrosis tubulointersticial. La fibrosis tubulointersticial se considera en general como la vía final común para la pérdida de la función renal en la nefropatía diabética. De hecho, la función renal y el pronóstico de la enfermedad podrían correlacionarse mejor con la fibrosis tubulointersticial que con los cambios glomerulares clásicos y tempranos. En general, se piensa que la acumulación de miofibroblastos activados es el principal contribuyente a la cicatrización renal progresiva en la diabetes. Estas células fibrogénicas pueden derivar de varias fuentes diferentes, incluida la transformación de fibroblastos residentes y células madre mesenquimales, el reclutamiento de fibroblastos de la médula ósea y la transdiferenciación tubuloepitelial a mesenquimal.
Neuropatía diabética
La neuropatía diabética involucra el sistema nervioso periférico y autonómico, afecta a casi la mitad de la población diabética. El riesgo de desarrollo de neuropatía diabética es directamente proporcional tanto a la duración como a la magnitud de la hiperglucemia. Además, algunas personas también pueden poseer facetas genéticas que influyen en su predisposición a desarrollar tales complicaciones. La prevalencia de la neuropatía diabética varía de un país a otro.
Los mecanismos de la vía de los polioles inducida por la hiperglucemia, la lesión de los AGE y el aumento del estrés oxidativo han sido implicados en su patogénesis. El daño a los nervios periféricos puede estar mediado por efectos sobre el tejido nervioso o por lesión endotelial con disfunción vascular. La neuropatía periférica en la diabetes aparece de varias formas según el sitio, manifestándose como neuropatía sensitiva, focal/multifocal y autonómica.

Existe una correlación significativa entre la neuropatía diabética y la existencia de una o más complicaciones macrovasculares que muestran que los pacientes diabéticos con neuropatía periférica, tienen tasas significativamente más altas de eventos cardiacos y enfermedad vascular periférica que los pacientes diabéticos sin neuropatía. La enfermedad cerebrovascular también es numéricamente más alta en DM con manifestaciones de neuropatía. Incluso, algunos estudios han demostrado una asociación entre la neuropatía y el desarrollo de retinopatía y microalbuminuria. También la neuropatía diabética autonómica cardiaca tiene una fuerte asociación con la retinopatía, neuropatía y control glucémico deficiente.
Alteraciones macrovasculares
Resistencia a la insulina y ateroesclerosis
Una proporción sustancial de pacientes diabéticos son obesos, la obesidad conduce a alteraciones en el metabolismo de los lípidos, disregulación de los ejes hormonales, estrés oxidativo, inflamación sistémica y distribución de grasa ectópica. El tejido adiposo es una fuente activa de mediadores inflamatorios y ácidos grasos libres (AGL). En consecuencia, los pacientes obesos con DM2 muestran niveles plasmáticos elevados de marcadores inflamatorios. Los AGL se unen al receptor Toll-like (TLR) activando NF-kB a través de la degradación del complejo inhibidor IkBα por IKKβ-cinasa. Como resultado, NF-kB desencadena la inflamación del tejido debido a la regulación positiva de los genes inflamatorios interleucina-6 (IL-6) y factor de necrosis tumoral alfa (TNF-α).
La activación del receptor Toll-like por AGL conduce a la fosforilación del sustrato-1 del receptor de insulina (IRS-1) por la cinasa amino-terminal c-Jun (JNK) y la PKC, alterando así su capacidad de activar a la baja los objetivos de fosfatidil inositol 3-cinasa (PI3-K) y Akt. Estos eventos moleculares dan como resultado la regulación negativa del transportador de glucosa 4 (GLUT-4) y, por lo tanto, la resistencia a la insulina (siguiente imagen). La regulación a la baja de la vía de PI3-K/Akt conduce a la inhibición de eNOS y a la producción disminuida de NO. Junto con la síntesis de NO reducida, la oxidación intracelular de AGL almacenado genera ROS que conduce a inflamación vascular, síntesis de AGEs, actividad de la PGI2 sintasa reducida y activación de la PKC (siguiente imagen).
La falta de señalización de la insulina en las plaquetas afecta la vía IRS1/PI3K, lo que resulta en acumulación de Ca++ y aumento de la agregación plaquetaria.
Trombosis y alteraciones de la coagulación en diabetes tipo 2
La diabetes tiene un mayor riesgo de eventos coronarios y mortalidad cardiovascular; este fenómeno se explica en gran medida por una disregulación de los factores implicados en la coagulación y la activación plaquetaria. Tanto la resistencia a la insulina como la hiperglucemia participan en la patogénesis de este estado protrombótico. La resistencia a la insulina aumenta el inhibidor del activador del plasminógeno-1 (PAI-1) y el fibrinógeno, y reduce los niveles del activador de plasminógeno tisular (t-PA) (como se observa en la siguiente imagen).

La hiperglucemia crónica y la resistencia a la insulina determinan una alteración significativa en los factores de coagulación, así como una mayor agregación plaquetaria, lo que lleva a un estado protrombótico. El aumento inducido por la diabetes de los niveles de factor tisular, activan la trombina convirtiendo el fibrinógeno en fibrina. La fibrina organizada aumenta aún más debido a altos niveles de PAI-1 y de t-PA reducidos. El aumento del contenido de Ca++, la estimulación de la trombina y la interacción con el factor de von Willebrand (vWF) a través del receptor de la glucoproteína IIb/IIIa, la cual provoca cambios en la forma de las plaquetas, la liberación de sus gránulos y la agregación. La liberación de micropartículas del endotelio lesionado y las plaquetas circulantes contribuyen a acelerar el desarrollo de un trombo. La disfunción endotelial precipita la ruptura de la capa endotelial y conduce a la exposición del colágeno y vWF, activando las plaquetas y favoreciendo la trombosis vascular.
Relación patogénica entre el daño microvascular y macrovascular en diabetes
La estrecha asociación entre la nefropatía diabética y la retinopatía es quizás el ejemplo mejor conocido por los médicos; el llamado síndrome del pie-ojo de la diabetes es otro. También se reconoce una estrecha asociación entre la disfunción eréctil y el riesgo cardiovascular, un vínculo que se cree que está mediado por una disfunción endotelial generalizada. Dado que los eventos patológicos tempranos son muy similares en los vasos pequeños y grandes, se ha formulado la hipótesis de que los cambios dentro de la microcirculación pueden impulsar el desarrollo y la progresión de la enfermedad de grandes vasos (como se esquematiza en la siguiente imagen).

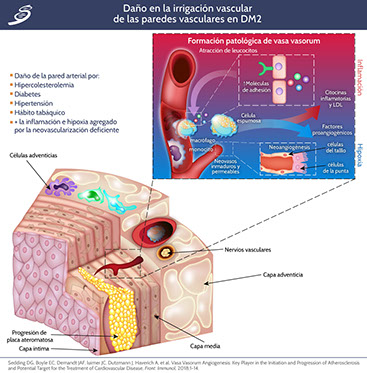
Los microvasos son la unidad funcional básica del sistema cardiovascular que comprende arteriolas, capilares y vénulas. Difieren de los vasos de mayor calibre (macrovasculatura) tanto en su arquitectura como en sus componentes celulares. En contraste con la macrovasculatura que suministra sangre a los órganos, los microvasos desempeñan un papel importante en el mantenimiento de la presión arterial y la entrega adecuada de nutrientes. La microcirculación también tiene sistemas reguladores que controlan la permeabilidad vascular y las respuestas miogénicas que pueden adaptar el flujo sanguíneo de acuerdo con las necesidades metabólicas locales. La alteración en la función microvascular puede surgir incluso antes de que se manifieste la hiperglucemia manifiesta y los cambios vasculares patológicos. La diabetes induce cambios patognomónicos en la microvasculatura, que afectan a la membrana basal capilar, incluidas las arteriolas en los glomérulos, la retina, el miocardio, la piel y los músculos al aumentar su grosor, lo que lleva al desarrollo de la microangiopatía diabética. Este engrosamiento finalmente conduce a una anormalidad en la función del vaso, lo que induce múltiples problemas clínicos como hipertensión, retraso en la cicatrización de la herida e hipoxia tisular. De manera similar, la neovascularización que surge de la vasa vasorum, puede interconectar la macro y microangiopatía, predecir la ruptura de las plaquetas y promover la aterosclerosis. El papel de la patología microvascular en las complicaciones diabéticas sistémicas, incluida la aterosclerosis macrovascular, sigue siendo un tema de debate adicional.
Neovascularización de vasa vasorum
La proliferación de vasa vasorum se asocia con un aumento del daño a la placa que promueve la aterosclerosis. Muchos procesos celulares como la inflamación, la perfusión de placa y la hemorragia intraplaca concomitante son críticos durante el desarrollo de placas ateroscleróticas y están relacionados con la proliferación de vasa vasorum. La neovascularización se desarrolla por el crecimiento tanto de la capa adventicia (hacia afuera) como de la luz arterial (hacia adentro) hacia la íntima. En la DM2, la rotura de la placa se asocia con un aumento de la angiogénesis, acelerando la aterosclerosis aún más con la microangiopatía por neovasculatura. La respuesta angiogénica inicial en el vasa vasorum adventicial, es un componente importante de los mecanismos homeostáticos, parece ser estimulada por la hipoxia a través de la identificación de aumento del factor inducible por hipoxia y del VEGF. El VEGF también contribuye a las complicaciones microvasculares al aumentar la permeabilidad vascular a las macromoléculas, la quimiotaxis de monocitos y la producción de factor tisular. La regulación al alza de VEGF también se informa en modelos experimentales de enfermedad renal diabética. Sin embargo, contrariamente a esto, se ha demostrado que el tratamiento con VEGF restablece la microcirculación en la vasa nervorum y limita la neuropatía diabética. En el ojo, un factor derivado del epitelio pigmentario de factor neurotrópico (PEDF) puede compensar la acción de VEGF por su potente inhibición angiogénica. El nivel de PEDF disminuye en la retinopatía proliferativa, mientras que los niveles de VEGF aumentan. La disminución de los niveles de PEDF también pueden contribuir a la nefropatía diabética. Otros factores de crecimiento como el factor 1 de crecimiento de tipo insulínico, el factor de crecimiento de fibroblastos básico y el factor de crecimiento de hepatocitos pueden fomentar la retinopatía proliferativa.

Bibliografía
Creager MA, Lüscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I, Circulation, 2003; 108(12):1527-32.
Paneni F, Beckman JA, Creager MA, Cosentino F. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: part I, Eur Heart J, 2013; 34(31):2436-43.
Vithian K. Microvascular complications: pathophysiology and management, Clinical Medicine 2010; Vol 10, No 5: 505–9
Wong TY, Gemmy Cheung CM, Larsen M, Sharma S y Simó R. Diabetic retinopathy, Nature Reviews Disease Primers, 2016; volume 2, Article number: 16012
Alicic RZ, Rooney MT, Tuttle KR. Diabetic Kidney Disease: Challenges, Progress, and Possibilities, Clin J Am Soc Nephrol, 2017;12(12):2032-2045.
Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, et al. Diabetic kidney disease, Nat Rev Dis Primers. 2015;1:15018.
Krentz AJ, Clough G, Byrne CD. Interactions between microvascular and macrovascular disease in diabetes: pathophysiology and therapeutic implications, Diabetes Obes Metab. 2007;9(6):781-91.
Chawla A, Chawla R, Jaggi S. Microvasular and macrovascular complications in diabetes mellitus: Distinct or continuum? Indian J Endocrinol Metab, 2016;20(4):546-51.
Fuller JH, Stevens LK, Wang SL. Risk factors for cardiovascular mortality and morbidity: the WHO Mutinational Study of Vascular Disease in Diabetes, Diabetologia, 2001;44 Suppl 2:S54-64.
Gross JL, de Azevedo MJ, Silveiro SP, Canani LH, Caramori ML, Zelmanovitz T. Diabetic nephropathy: Diagnosis, prevention, and treatment, Diabetes Care, 2005;28:164–76.
Soedamah-Muthu SS, Chaturvedi N, Witte DR, Stevens LK, Porta M, Fuller JH. EURODIAB Prospective Complications Study Group. Relationship between risk factors and mortality in type 1 diabetic patients in Europe: The EURODIAB prospective complications study (PCS), Diabetes Care, 2008;31:1360–6.
Hovind P, Tarnow L, Rossing P, Jensen BR, Graae M, Torp I, et al. Predictors for the development of microalbuminuria and macroalbuminuria in patients with type 1 diabetes: Inception cohort study, BMJ, 2004;328:1105.
Jarman S. Nerves Need Blood: Vasa Nervorum, Classical Osteopathy in Ontario [january 2, 2018] disponible en: https://classicalosteopathyontario.wordpress.com/2018/01/02/nerves-need-blood-vasa-nervorum/ [revisado septiembre 2018]
Sedding DG, Boyle EC, Demandt JAF, luimer JC, Dutzmann J, Haverich A, et al. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease, Front. Immunol, 2018;1-14.
Slevin M, Krupinski J, Badimon L. Controlling the angiogenic switch in developing atherosclerotic plaques: possible targets for therapeutic intervention, J Angiogenes Res, 2009;1:4.
Aviso de privacidad
Diseño: A. Victoria Pérez