__
__
__
EDUCACIÓN MÉDICA
DEMENCIA VASCULAR
El concepto de demencia causada por una patología cerebrovascular ha evolucionado considerablemente a lo largo de los años. Durante muchas décadas, la demencia vascular se atribuyó a la esclerosis de las arterias cerebrales que causaba lesión isquémica difusa y atrofia cerebral. La primera desviación significativa de este concepto, inspirada por el trabajo de Tomlinson y sus colegas, fue propuesta por Hachinski, quienes sugirieron que la demencia, en las bases vasculares, fue causada por lesiones isquémicas múltiples y discretas en pacientes con factores de riesgo vascular, como la hipertensión (demencia secundaria a multiinfartos), planteando la posibilidad de prevenir las enfermedades cerebrovasculares y por lo tanto, la demencia.
FISIOPATOLOGÍA
Las células de la unidad neurovascular participan en la iniciación y expresión de respuestas inmunes adaptativas e innatas del cerebro. Los pericitos y los macrófagos perivasculares tienen potencial para la presentación de antígenos, el primer paso en la inmunidad adaptativa; mientras que las células endoteliales y las células de la microglia están ricamente dotadas de receptores de inmunidad innata que incluyen CD36, receptores de toll-like (TLR) y el receptor para avances en productos finales de glicación (RAGE). El espacio perivascular, que drena al espacio subaracnoideo y luego a los ganglios linfáticos cervicales, es el "brazo aferente" a través del cual los antígenos cerebrales alcanzan el sistema inmune sistémico. Las células de la unidad neurovascular también regulan el "brazo eferente" del sistema inmune, que depende de la transferencia de células inmunes efectoras al cerebro. En condiciones de hipoxia-isquemia, las células endoteliales expresan receptores de adhesión, tales como P-selectina, E-selectina, ICAM y VCAM, que son fundamentales para la transferencia de leucocitos circulantes al espacio perivascular. Al mismo tiempo, se requiere la comunicación con macrófagos perivasculares, ubicados en contacto con la membrana basal vascular, para que las células inflamatorias, como los linfocitos, crucen la membrana glial y se muevan desde el espacio perivascular hacia el parénquima cerebral. Los astrocitos expresan ligandos de "muerte" (CD95L) en sus extremidades perivasculares y controlan el tráfico inmune desencadenando la apoptosis de los linfocitos CD95+ que intentan ingresar al cerebro. Por lo tanto, la unidad neurovascular es un importante punto de control que regula los brazos aferentes y eferentes del sistema inmune y conforma la respuesta inmune del cerebro. La homeostasis vital para la vascularización, sucede mediante células progenitoras endoteliales circulantes (EPC, por sus siglas en inglés), células madre hematopoyéticas implicadas en el mantenimiento y células endoteliales que se encuentran en constante reparación. El desarrollo y la función de EPC está controlado por células T CD31 + (células T angiogénicas) a través de las citocinas proangiogénicas de liberación. Por lo tanto, las células inmunes también están involucradas en el mantenimiento de la homeostasis vascular.
Debido a su ubicación en el borde distal entre diferentes territorios vasculares y a la susceptibilidad de su vasculatura a los factores de riesgo, los tractos de sustancia blanca profunda son particularmente vulnerables a la insuficiencia vascular. Incluso en individuos sanos, un estado de hipercapnia, lo cual tiene un efecto vasodilatador potente, no aumenta, pero reduce en la sustancia blanca periventricular, lo que sugiere que la vasodilatación de vasos sanguíneos desvía el flujo sanguíneo a otras regiones (robo intracerebral). Este hallazgo destaca la precariedad hemodinámica de la sustancia blanca periventricular, incluso en ausencia de daño vascular. La creciente evidencia sugiere que el suministro de sangre cerebral a la sustancia blanca se ve comprometida en el/los infarto(s) cerebrovascular(es), es por ello que el flujo de reposo se reduce en áreas de leucoaraiosis y la reactividad vascular se atenúa. La autorregulación cerebrovascular está alterada, lo que aumenta la susceptibilidad de la sustancia blanca al daño durante la fluctuación de la presión arterial, teniendo también un papel importante la reducción del flujo sanguíneo que puede contribuir al daño de la materia blanca, determinándose que la reducción de la circulación cerebral puede observarse antes del inicio de la demencia. Debido a su vulnerabilidad hemodinámica, las regiones profundas de la sustancia blanca están marginalmente perfundidas y, en presencia de factores de riesgo vascular, sus vasos pueden ser incapaces de adaptar la corriente sanguínea a las necesidades metabólicas del tejido. De acuerdo con esta hipótesis, los estudios post mortem han demostrado que las áreas de leucoaraiosis son crónicamente hipóxicas, como lo indica la expresión de factores y genes relacionados inducibles por hipoxia.
Además de los factores locales que afectan los microvasos de la sustancia blanca, también intervienen factores sistémicos de acción más amplia. Las lesiones de la materia blanca y los accidentes cerebrovasculares lacunares se asocian con aumentos en los niveles circulantes del inhibidor de óxido nítrico-sintasa dimetilarginina asimétrica (ADMA). ADMA puede contribuir al deterioro de la vasodilatación dependiente de ON en arterias periféricas y cerebrales. Estos hallazgos implican la pérdida de la elasticidad de vasos sanguíneos grandes, como arterias, y el aumento del estrés pulsátil en microvasculatura, especialmente aquellos que se ramifican directamente del polígono de Willis, en el daño microvascular que subyace a las lesiones de materia blanca. Cambios microvasculares similares ocurren también en otros órganos, lo que sugiere que la enfermedad de vasos pequeños en el cerebro puede ser la manifestación de una vasculopatía sistémica. La evidencia revisada anteriormente sugiere una convergencia de factores patógenos en los vasos sanguíneos cerebrales, lo que a su vez conduce al daño de la sustancia blanca. La disfunción endotelial inducida por el estrés oxidativo causada por factores de riesgo, es muy probablemente un evento temprano que conduce al daño de la materia blanca. La disfunción endotelial tiene dos consecuencias patogénicas principales: reducción en la perfusión del flujo sanguíneo cerebral de la sustancia blanca marginal, y alteraciones en la permeabilidad de la barrera hematoencefálica (BHE).
La hipoperfusión y la alteración de la BHE conducen a un estrés oxidativo adicional al inducir hipoxia tisular y extravasación de proteínas plasmáticas como fibrinógeno. El edema tisular resultante del aumento de la permeabilidad de la BHE puede exacerbar estas alteraciones al comprimir los vasos sanguíneos y reducir aún más el flujo sanguíneo cerebral. La hipoxia tisular y el estrés oxidativo activan las vías inflamatorias a través de la transcripción dependiente de factor nuclear kappa-beta (Nf-κβ), que conduce a la producción de citocinas y moléculas de adhesión en células endoteliales, astrocitos reactivos y microglia activada. La hipoxia, la inflamación y el estrés oxidativo dañan oligodendrocitos y conducen a un desacoplamiento trófico en la unidad neurovascular que, a su vez, contribuye al daño de las células vasculares y los oligodendrocitos. En el proceso fisiopatológico de la demencia vascular, diversas moléculas juegan un papel importante que se observa en el siguiente esquema.
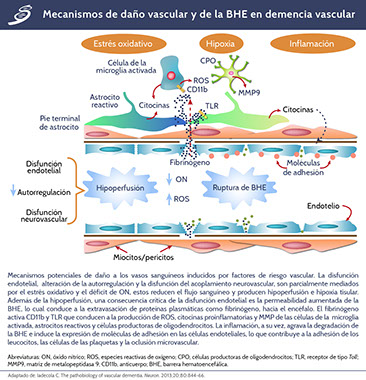
El daño a oligodendrocitos, el estrés oxidativo y la inflamación conducen a la desmielinización e intentos de remielinización a través de la proliferación de OPC. La detención del desarrollo de células progenitoras de oligodendrocitos, debido al exceso de productos de degradación de hialuronano, conduce a la acumulación de estas células que secretan metaloproteinasa 9 (MMP9, por sus siglas en inglés) empeorando el deterioro. Una vez que se produce la desmielinización, el aumento en el requerimiento de energía por parte de los axones desnaturalizados, agrava el estrés hipóxico del tejido, dando lugar a un círculo vicioso que perpetúa estos procesos patogénicos y exacerba el daño tisular.
Bibliografía
1. Adaptado de: Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;20;80:844-66
2. O'Brien JT, Thomas A. Vascular dementia. Lancet. 2015; 24;386(10004):1698-706.
3. Barbay M, Taillia H, Nedelec-Ciceri C, et al. Vascular cognitive impairment: Advances and trends. Rev Neurol (Paris). 2017;173:473-480.
Aviso de privacidad
Diseño: A. Victoria Pérez